Get Complete Project Material File(s) Now! »
ntroduction to Phylogenetics and Phylogenomics
In the mid-1800s, Darwin’s theory of the origin of species gave birth to the field of evolution. Ernst Haeckel, a German zoologist also came up with a sketch which became a blueprint to what we know today as a phylogenetic tree. At that time, evolutionary relationships were built upon the similarities between specie morphology. It was assumed that sharing of common phenotypic traits might indicate a common ancestry of organisms represented in a tree by branches joined by an intermediate node. In Figure 1.1, the branches outlined in purple represent the species tree. As time progressed and the genetic basis of life was generally recognised, species comparison evolved into gene sequence comparisons, leading to gene trees. Gene trees (Figure 1.1 in blue, red and green) do not always agree with species trees owing to events such as horizontal gene transfer (HGT), gene duplication and an uneven rate of evolution of different genes (Figure 1 blue to green and red).
This sometimes led to conflicting predictions of speciation events (Lin et al., 2011; Swenson and ElMabrouk, 2012; Bezuidt et al., 2016). Nowadays, phylogenetics is used in many aspects of biology. These fields include analysis of relationships between species (Takahashi et al., 2001; Zhaxybayeva et al., 2006), improvement of methods utilising annotation information such as paralogues (Finnerty et al., 2009; Berendzen et al., 2012; Chai et al., 2014), population evolution discovery (Francois and Mioland, 2007; Li and Durbin, 2011), pathogen and cancer studies in the field of medicine (Cawley and Talbot, 2006; Stecher et al., 2013) and detection of HGT through phylogenetic tree comparisons (Poptsova and Gogarten, 2007). Phylogenetics and its toolset are becoming ever more important, as applying it is a vital skill in supporting other type of studies, such as metagenomics (Filipski et al., 2015).
Gene-based Phylogenetics
A phylogenetics study includes many steps, of which the final aim is to create a phylogenetic tree consisting of branches denoting the relationship of common ancestry between each species in the study. Phylogenetic tree construction is divided into two main types of methods, consisting of character-based and distance-based approaches. For distance-based approaches, as the name states, the sequence comparison between two species is calculated as a number of weighted evolutionary events estimated by a certain criterion or algorithm. These distances, which denote the diversity between species, are then used in a tree construction algorithm such as neighbour joining (NJ) (Saitou and Nei, 1987) to resolve the final phylogeny. Character-based approaches, on the other hand, look at alignments of all sequences simultaneously and consider every single character difference along all possible (or plausible in the case of heuristics) tree topologies as a likelihood penalty with the aim to identify the most likely tree topology. Based on the different methods, the best tree is chosen upon a tree score for which each method has its own selection criteria. For example, maximum parsimony considers the smallest number of single-character substitutions between aligned sequences as the most likely tree path. Maximum likelihood (ML) considers the log likelihood score based on a chosen substitution model, and the Bayesian method the best posterior probability (Yang, 1996; Yang and Rannala, 1997).
Approaches to Phylogenomics
As explained in the introductory section, phylogenomics was derived from phylogenetics to cover its shortfalls in handling large amounts of sequencing data in larger regions produced from NGS technologies (Chan and Ragan, 2013). Because of this fact, some phylogenomic approaches are very similar to phylogenetics, with some phylogenomics tools being upgraded versions of current phylogenetics toolsets. However, for the relevance of this section, we took a more in-depth look at other phylogenomic methods, which take different approaches. These include supermatrix and supertree methods, average nucleotide identity (ANI), genome BLAST sequence phylogeny, pangenomic analysis of clusters of orthologous genes, multi-locus sequence typing (MLST), alignment-free compositional algorithms and whole genome alignment. We will also discuss each method in detail, as well as its relevance in terms of current phylogenomic research and the tools that use these approaches. Finally, the problems of phylogenomics in the current context and the pros and cons of each method are evaluated.
Ortholog-based Approaches
One commonly used phylogenomic method is comparing the distribution of orthologous genes in genomes. The presence and absence of genes can determine the similarities of different taxonomic units. Techniques for identification of orthologous genes have been proposed by a reciprocal BLASTP alignment of translated complete DNA sequence (CDS), by complete genome alignment, or by combinatorial approaches (Sims et al., 2009). Efficient Database framework for comparative Genome Analyses using BLAST score Ratios (EDGAR) is a good platform that can identify orthologs using comparative analysis (Blom et al., 2009). This platform contains a large database containing orthologs from over 500 genomes across 75 genera in the National Centre for Biotechnology Information (NCBI) database. Orthologs in this case are defined under a strict criterion as genes with conserved function and diverged from a speciation event (Fitch, 1970). Hence, based on ortholog comparison, one can identify evolutionary events through speciation. Alignment and Annotation Free Methods Another innovative approach of CVTree was the reduction of the background noise resulting from an assumption of context-independent substitution of residues in protein sequences.
This is done by considering amino acids as part of evolutionary stable K-mer oligopeptides shaped and governed by natural selection pressure, which is achieved by applying the Markov chain model (Brendel et al., 1986). The K-string oligopeptides with non-zero difference between the observed K-string frequency and estimated frequency calculated based on frequencies of K-2 substrings were used for the construction of genome-specific compositional vectors. These compositional vectors were the key elements of measuring the evolutionary distances between genomes. The similarity measure consists of calculating the correlation between two compositional vectors estimated for given genomes. Finally, the correlation values were converted into distance values ranging from 0 to 1 and the distance matrix was processed by the NJ method to plot the final phylogenetic tree. The first limitation of this method is that it translates sequences of protein-coding genes instead of whole genome information.
Table of Contents :
- Declaration
- Plagiarism Statement
- Abbreviations
- List of Figures
- List of Tables
- List of Supplementary Figures and Tables
- Summary
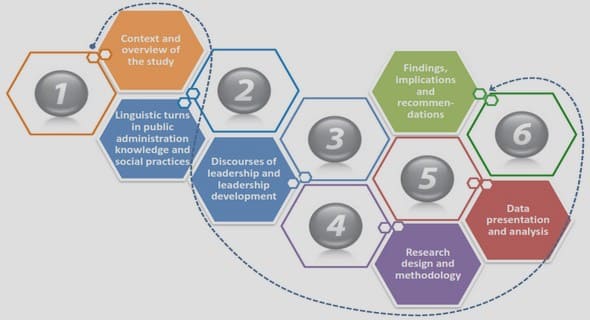
- Chapter 1. Literature Review
- 1.1 Introduction to Phylogenetics and Phylogenomics
- 1.2 Current Methods and Approaches to Phylogenetic Inferences
- 1.2.1 Gene-based Phylogenetics
- 1.2.2 Approaches to Phylogenomics
- 1.2.2.1 Supermatrix and Supertree-based approaches
- 1.2.2.2 Approaches to Phylogenomics
- 1.2.2.3 Sequence Alignment Approaches (MAUVE)
- 1.2.2.4 Alignment and Annotation Free Methods
- 1.3 Application of Nucleotide K-mers in Phylogenetics and Concept of Oligonucleotide Usage Pattern
- 1.4 Tree Comparison and Model Evaluation
- 1.5 Aims of Current Project
- Chapter 2. Analysis of Possible Evolutionary Forces Shaping Oligonucleotide Usage Patterns
- 2.1 Introduction and Theory Overview
- 2.2 Relations Between OUP and Codon Usage in Bacteria Genomes
- 2.2.1 Selection of Bacterial Genomes for Case Studies
- 2.2.2 Analysis of Emission Patterns Calculated for Different Groups of Microorganisms
- 2.3 Program Modelling of Context and Codon Dependent Genome Evolution
- 2.4 Discussion
- Chapter 3. Creation and Comparison of OUP-based Tree to Common Phylogenetic Inferences
- 3.1 Methods Used for OUP Calculation and Comparison
- 3.2 Selection of Taxonomic Groups for Case Study
- 3.3 Methods used for the Construction of Phylogenetic Trees by Alignment-based and Alignment-free Approaches
- 3.4 Evaluation of the OUP Based Algorithm by Comparison of Resulting Trees
- 3.4.1 Comparison of OUP Inferences to Other Genome-based and Gene-based Phylogenetic Trees
- 3.4.2 Resolving Phylogenetic Relations between Prochlorococcus Strains by OUP Approach
- 3.4.3 Testing of the OUP Approach on Artificial Sequences Simulating Speciation Events
- 3.4.4 Bootstrapping Test for the Consistency of OUP Approach based on the Variation in Lengths
- 3.5 Reconciliation of Tree Topologies by Logistic Functions
- 3.6 Discussions
- Chapter 4. Design and Implementation of the Program SeqWord Phylogenomics
- 4.1 SWPhylo Algorithm of OUP-based Phylogenetic Inferences
- 4.2 Design of the Web-based Software Tool SWPhylo
- Chapter 5. Conclusions
- Acknowledgement
- References
- Appendix
GET THE COMPLETE PROJECT
Mathematical modeling of evolutionary changes of oligonucleotide frequency patterns of bacterial genomes for genome-scale phylogenetic inferences