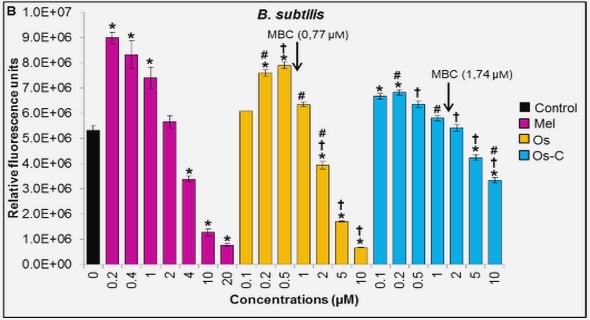
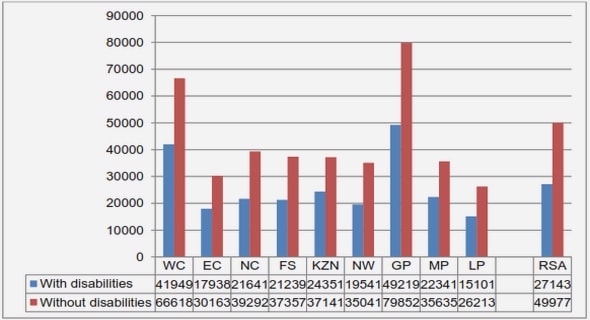
Get Complete Project Material File(s) Now! »
Electronic structure methodology
Introduction
The morphology of the gold core, the nature of the ligands, their mode of grafting on the metal surface and the presence of dopants have been analyzed in recent years showing the significant impact these parameters have on the catalytic and optical properties of gold nanoclusters (GNC). [1-5] ESI and IMS mass spectrometry, UV – Vis and FT – IR spectroscopy and X – Ray diffraction characterization techniques provided important information for elucidation of GNC structures and their properties. [6-12] However, because of their ultra – small size, the determination of the structure and the study of the properties of ligand capped GNCs remains a challenge. The ligand organization on the gold surface, for instance, cannot be easily determined experimentally because of the small sizes (of the order of 1 – 2 nm) however, it could be used as advantage to tune the optical properties of GNCs.[13-15] For these reasons, computational chemistry and electronic structure methods play a complementary role to the experimental characterizations in the understanding of the electronic structure of GNCs and its impact on their structural and optical properties.
GNCs have a large number of atoms and electrons. The computation of their electronic structure by wave function based quantum chemistry methods is very demanding. DFT methods based on the electron density require to treat only 3 electronic variables at a given configuration of the nuclei compared to the 3N for wave function methods (where N is the number of electron of the system). Solid state DFT implementations were extensively used to investigate GNCs.[12, 16-27] Recently, thanks to the significant increase in computer resources, mainly memory, it became possible to compute the electronic structure of GNCs (ground and excited electronic states) and to determine their equilibrium geometry using the Kohn – Sham [28, 29] implementation of DFT in quantum chemistry. This is the methodology we adopted in this thesis. The Kohn – Sham implementation implies to use 3N electronic coordinates. Its cost is basically equivalent to that of a Hartree – Fock computation (that is to say, one determinantal wave function) when hybrid functionals are used. The use of pure functionals can considerably speed up the computation since only one electron integrals need to be computed.
A further challenge is that the gold atom has a heavy nucleus. Therefore, the electrons in gold (and in general in post – lanthanide elements) experience a strong nuclear field, and move at velocities comparable to that of light. For this reason, relativistic effects become important and have an impact on the chemical properties,[30] structure,[31] catalytic activity and electronic structure [32]. Relativistic effects are also responsible for weak “’aurophilic’” interactions. [32-35] Due to the relativistic effects the 6s outer shell of Au undergoes a contraction which leads to a decrease in the 5d – 6s gap. As a consequence, there is a larger s – d hybridization in gold. [34]
Including relativistic effects in quantum chemistry computation is a very challenging task, even for atoms. Moreover, the computation of the electronic structure and properties of heavy elements compounds represents a considerable computational cost due to their large number of electrons. The use of effective core potentials represents a good approximation for transition metal compounds. It allows to both reduce the number of active electrons to the valence electrons and to take relativistic effects into account. When using effective core potentials, the chemically inert core electrons are replaced by an effective potential and the relativistic effects on the outer valence electrons are included, leading to a more feasible calculation.
Another problem to face is the description of the ligand shell and the ligand – Au interface. Noncovalent interactions and dispersion forces play an important role on the stability of gold – organic complexes [39] and also on their structure [40, 41]. Among the different computational approaches, many determinant wave function quantum chemistry methodologies, such as Coupled Cluster (CCSD(T)), provide an accurate description of non – bonding van der Waals interactions. Unfortunately, the use of this method it is not feasible for the nanoclusters studied here because of their size (number of electrons and number of nuclear degrees of freedom). An alternative is to use a perturbative approach, such as MP2 (Møller – Plesset perturbation theory) that has been employed in several investigations as a reference for DFT studies where different functionals are tested [39, 40, 42] since MP2 offers a good description of dispersion forces for the ground state. Theoretical studies on bare and protected small gold clusters have shown that computations using the long – range corrected CAM – B3LYP functional provides a good agreement with MP2.
Therefore, here, the quantum chemistry Kohn – Sham implementation of Density Functional Theory is used to study the effect of the metal core structure, of the chemical nature of the ligand on the gold surface and also of the impact of two different ligands protecting a metal core on the electronic structure and optical properties. All computations on the bare and protected gold clusters were carried out using the Gaussian 09 program.[44] The long – range corrected functional CAM – B3LYP [36] was used together with the LANL2MB relativistic corrected pseudopotentials and basis sets [37, 38] for the gold atoms and the 6 – 31G(d) Gaussian basis set for the ligand shell. Natural population analysis and natural bond orbital methodologies [45, 46] were used to compute the partial charges on the atoms of the clusters. Contributions from different fragments (gold core and the ligands) to the frontier molecular orbitals and the density of states (DOS) were obtained with AOMix program [47] using the default MPA (Mulliken population analysis). The excited states and the UV – Vis spectra were computed at the equilibrium geometry of the ground electronic state with the linear response TD – DFT method as implemented in Gaussian 09.
In this chapter, first, we describe the foundations of ground state Density Functional Theory followed by a brief explanation of the Berny algorithm implemented by H. Bernhard Schlegel [48] used to compute the equilibrium structure of the ground state of the gold clusters. Then, the description of the computation of thermodynamic quantities such as ΔG, ΔH and ΔS for ligand binding is outlined. Also, a brief overview of charge distribution computation [45, 46] is provided. In order to ensure the charge distribution trends and since no absolute definition of atom charge has been established, we compared the results of Natural population analysis with those of the CHelpG [49] and the M – K [50] charge distribution approaches.
The algorithm used in this work to find the equilibrium geometry of the GNCs is called Berny [48] algorithm in redundant internal coordinates [62-64] using GEDIIS (geometry optimization method using an energy – represented direct inversion in the iterative subspace algorithm)[65] and it is implemented in Gaussian 09. Here is a brief explanation about how the algorithm finds the equilibrium geometry :
1) Having our guess of the equilibrium structure and considering that the KS orbitals are obtained already from the self – consistent procedure and having in mind that, analytic first derivatives are available for almost all density functionals,[66] the electronic energy, nuclear repulsion and the potentialcan be calculated together with a first estimation of the Hessian matrix by using connectivity derived from atomic radii and a simple valence force field.
2) The first Hessian[ matrix]=guess is improved[ ]= at− each point by calculating the gradient. Notice that we are looking for and , where F stands for the force the nuclei feel due to the potential.
3) The trust radius is updated, that is to say, the maximum size for the optimization step. [68]
4) If a minimum is detected a 5th or 4th order polynomial is used to fit the line between the initial point and that which could be the new configuration (or already the minimum). To ensure that the polynomial has an only one minimum the fit is subjected to the constraint that the second derivative of the polynomial just reach zero at its minimum. [52]
5) Maximum force component and maximum step component are used as convergence criteria to evaluate the new nuclear configuration. The step is the change between the most recent point and the next to be computed.
6) If the new configuration does not reach the convergence criteria, the midpoint of the line connecting the new and the best previous point is taken as a new configuration.
7) All the optimization steps apply themselves until a minimum is localized within the convergence criteria.
Thermochemistry and vibrational analysis
Computation of thermochemical quantities
The computation of thermodynamic quantities such as ΔG, ΔS and ΔH allows us to calculate the binding energy of a molecule, compare the stability of different system configurations and to determine the free energy and enthalpies of formation of a specific reaction product.
The contributions to the canonical partition function (q), total internal thermal energy ( ), entropy (ΔS) and heat capacity (Cv) from the vibrational, rotational, electronic and translational motion are calculated for the ground electronic state assuming an isolated molecule and assuming that the first electronic excitation energy is much larger than (where is the Boltzmann constant and T is the temperature in K).
Computation of normal mode frequencies
The Hessian matrix (see Eq. 26) allows us to compute the normal mode frequencies and force constants. This gives us information about the strength of a bond. The next steps summarize how the normal mode frequencies are calculated:
1) First we have to be sure that the first derivative of the energy respect the atom displacement is zero, that is to say, the geometry of the molecule corresponds to point where the forces on the nuclei are zero.
2) Conversion of the force constants calculated′ by the Hessian matrix to a mass weighted coordinates is performed. The new Hessian matrix,can be expressed as: ′ = √ (35)
Here, and are the mass of the atoms corresponding to the set of coordinates and , respectively, at the equilibrium geometry.
3) Determination of principal axes of inertia, separation of rotation and translation to only analyze the 3M – 6 or 3M – 5 vibrations is performed (M being the number of nuclei). Finally, transformation of the Hessian matrix to internal coordinates is done. More details about these three calculations can be found by consulting reference [70].
4) Diagonalization of yields the 3M eigenvectors and 3M eigenvalues . The Hessian eigenvectors are called normal modes and the squared root of the eigenvalues ( / ) provide the frequencies of the normal modes ( ), as can be seen in the next equation
As mentioned in the geometry optimization section, if all the calculated frequencies are real, we are at the global minimum or at a saddle point (secondary minimum on the PES). If some of the frequencies are imaginary, we are at a transition state.
Charge distribution computation
Calculation of atomic charges could be very helpful for the understanding of the grafting on ligand – metal systems. Depending on the nature of the ligand (whether if it is electron donor or acceptor) and by knowing the atomic charges on the metal surface, one is able to predict the preferred sites for a ligand to interact. Moreover, it has been shown that the charge distribution on hybrid metal – organic systems have a significant impact on their optical [13] and surface properties.
However, this method does not show any convergence while increasing the size of the basis set. The assignment of half the overlap population to each basis function (see Eq. 40) is arbitrary and it can actually give chemically meaningless results, as occupation numbers larger than 2 or with negative values. Therefore, here we use Natural Population Analysis (NPA) [45, 46] to obtain the natural partial charges based on a orthonormal set of natural bond orbitals (NBO).
As mentioned at the beginning of this section, there is not an absolute definition of partial charges, hence it is important to be sure that we can rely on our results. Some studies on ligand capped GNCs show that by using different approaches to compute the partial charges; NBO, M – K, AIM and CHelpG, for instance, the same trends are found. [43, 69] The main difference between NBO and the other two approaches is the fact that NBO scheme is based on localized natural atomic orbitals while the M – K and the CHelpG methods are computed to be consistent with the molecular electrostatic potential (MEP) which is an observable. A molecule can be seen as a group of charged points (nuclei) smeared out into a continuous distribution. In quantum chemistry the MEP of a molecule at point 1 .
Time dependent DFT
DFT is first used to describe the ground electronic state of a system. However, by applying a weak time – dependent oscillating perturbation to the ground state we can obtain information about the excited states. This approach is called time – dependent density functional theory (TD – DFT). Excitation energy and oscillator strengths can be obtained allowing us to compute the UV – Vis spectrum. In TD – DFT, since we are analyzing the response of the ground state of our system to a weak time – dependent oscillating perturbation the time – dependent Schrödinger equation has to be solved.
The theoretical basis of TD – DFT [75] was developed by Runge and Gross. The Runge – Gross theorem states that there exist a one – to – one mapping between time – dependent external potential and the time – dependent densities. [76] In other words, the expectation value of any operator is a unique functional of the time – dependent density. As in the time – independent case, the Runge – Gross theorem for a non – interacting system leads to a Kohn – Sham (KS) (now) time – dependent equations (in atomic units).
Linear – response TD – DFT is the formalism used to obtain excitations energies. First, as usually done in perturbation theory, we obtain the energy and the electron density for the unperturbed time – dependent case, then, the perturbation is applied.
Solvation. The Polarizable – continuum model (PCM)
Most of our theoretical work was performed in the gas phase. However, in chapter VI the computation of the 13C NMR spectra of the set of (NHCa)n – Au38 complexes, the fully ligated Au38(NHCa)9 model cluster and of the bis(NHC) AuI complex (NHCa being 1, 3 – dimethyl – 1H – benzimidazolium; an n – heterocyclic carbene ligand) were carried out at the DFT / CAM – B3LYP level. It is well known that the signals in a NMR spectrum are significantly affected by the chemical environment. Therefore, the effect of the solvent (d6 – DMSO) was taken into account on the equilibrium structure as well as in the NMR spectra computation by using the polarizable – continuum model (PCM).
The PCM was developed more than 30 years ago and it still represents an attractive alternative to explicitly models of the solvent effect in a molecular system.
In section 2.4 in this chapter, a molecule is assumed to be a group of charged points (nuclei) smeared out into a continuous distribution. Here, is the solute charge distribution and is the general permittivity. The general permittivity takes the value of 1 when falls in the cavity representing the solute (molecule). is the electric potential produced by the charge distribution of the molecule (solute) .
The cavity in Gaussian 09D is generated via a set of overlapping spheres: each nucleus in the the solvent. ⃗ solute molecule is surrounded by a sphere of radius 1.2 times the van der Waals radius of that atom.
Hybrid QM – MM methodology
The size and the complexity of a molecule together with the process under study play a crucial role on the choice of computation methodology. DFT and ab – initio approaches provide valuable information on chemical reactions energetics and charge transfer process, for instance. However, for some large systems, for example, macromolecules, such as enzymes or proteins, the use of (quantum mechanics) QM can be extremely demanding computationally and very long, in other words, prohibitive. We have then to compromise on the accuracy of our calculations by using a lower level method. An alternative is to use molecular mechanics (MM) but MM typically does not describe the formation and rupture of bonds in a chemical reaction.
At this point, hybrid QM – MM approach can be very helpful. The basic idea proposed by Warshel and Levitt [82] in 1976 is to divide the whole system in several portions treated with different levels of theory, QM in the most important region and MM for the rest, as exemplified in figure 1. More than 2 layers can be defined since the QM regions can be treated with different QM levels of theory. [83]
In our case, the ligand layer can represent a large number of atoms therefore, QM – MM has been used in few cases only to obtain an equilibrium structure close to the full QM operator only depends on the nuclear coordinates and is written:
The subscript “i” and “j” in Eq. 65 run on the electron coordinates while “α” and “β” refer to the nuclei. The mass of electrons and nuclei are and , respectively. The vectors and stand for the nuclei position in space while and stand for the electrons. The nuclear charges are expressed as . The first and second term stand for the electrons and nuclei kinetic energy. The third one and the fourth represent the nuclear repulsion and nuclear – electron attraction, respectively. Finally, the fifth and last term in the molecular Hamiltonian is the electron̂– electron repulsion.
Partitioning of a system into different regions studied by a hybrid potential. There are 2 regions in this representation. The QM (in the center) which corresponds to high – level quantum – mechanical calculations while the dark green area, MM, is modeled with molecular mechanics force fields.
Principles of the QM – MM method
As mention already in section 2.1 (Eq. 1), a non – relativistic molecular system considering time – independent interacting particles can be described by solving the time – independent Schrödinger equation, now taking into account the different regions: QM – MM.
Table of contents :
Chapter I. Introduction
References
Chapter II. Electronic structure methodology
Introduction
2.1 Density Functional Theory
2.2 Equilibrium geometry determination and the Berny optimization algorithm
2.3 Thermochemistry and vibrational analysis
2.3.1 Computation of thermochemical quantities
2.3.2 Computation of normal mode frequencies
2.4 Charge distribution computation
2.5 Time dependent DFT
2.6 Solvation. The Polarizable – continuum model (PCM)
2.7 Hybrid QM – MM methodology
2.7.1 Principles of the QM – MM method
2.7.2 The QM – MM junction
2.8 References
Chapter III. Charge redistribution effects on the UV – Vis spectra of small ligated gold clusters
Introduction
3.1 Publication
3.2 References
Chapter IV. Geometry , electronic structure and optical properties of the [Au25(SR)18]− (R = CH3, TP,
SPhPh, SPhPhPh, 4ATP) and the Janus [Au25SR’xSR’’18 – x]− R’ = Ph and R’’ = 4ATP, CH3(CH2)4COOH)
clusters
Introduction
4.1 The monoligand protected [Au25(SR)18]− cluster
4.1.1 Structure and partial charge distribution of the monoligand protected [Au25(SR)18]–
4.1.2 Optical properties
4.2 The Janus [Au25SR’xSR’’18 – x]– cluster R’ = Ph TP and R’’ = 4ATP, CH3(CH2)4COOH)
4.2.1 Structure and partial charge distribution of the Janus [Au25(TP)x(S(CH2)5COOH)18 –x]–
4.2.2 Structure and partial charge distribution effects of thiolate ligands on the Janus
[Au25(TP)x(4ATP)18 – x]– cluster
4.2.3 Optical properties
4.3 Conclusion
4.4 References
Chapter V. Experimental approach to gold nanocluster synthesis and characterization
Introduction
5.1 Synthesis and characterisation of the Au25(TP)18 and Au25(4ATP)18 nanoclusters
5.1.1 Synthesis protocol of the Au25(TP)18 and Au25(4ATP)18 nanoclusters
5.1.2 Characterization of the Au25(TP)18 and Au25(4ATP)18 nanoclusters
5.2 The Au25(SR)18SR’)18 – x cluster synthesis protocol by simultaneous ligand addition
5.2.1 Synthesis protocol of the Au25(SR)18SR’18 – x nanoclusters
5.2.2 Characterization of the Au25(SR)18SR’18 – x clusters
5.3 Conclusions
5.4 References
Chapter VI. Experimental and theoretical study of the reactivity of gold nanoparticles towards benzimidazole – 2 – ylidene ligand
Introduction
6.1 Publication 2
6.2 References
Chapter VII. Dynamics of phosphine and 1, 3 – dimethyl – 1H – benzimidazolium ligand on gold nanocluster surface
7.1 Introduction
7.2 Structure and natural charge distribution
7.3 Optical properties
7.4 Site reactivity of the PH3 on the Au38 cluster surface. Comparison with NHC
7.5 Conclusions
7.6 References
Conclusions and Perspectives
References
Appendices
1. Supporting information: Charge Redistribution Effects on the UV-Vis Spectra of Small Ligated Gold
Clusters: a Computational Study. (Chapter III)
2. Supporting information: Experimental and Theoretical Study of the Reactivity of Gold
Nanoparticles towards Benzimidazole-2 – ylidene Ligands. (Chapter VI)