Get Complete Project Material File(s) Now! »
PEMFC Overview
Principle
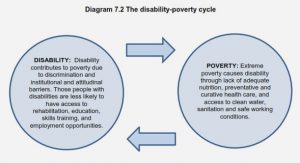
In batteries, electrochemical reactions are used to generate electricity, and the reacting materials, as well as the reaction products, are typically metals or other solid compounds (oxides, salts, etc.). In contrast, in fuel cells, both the reactants and the products are liquids or gases. This permits a continuous supply of reactants to the cell and a continuous removal of the reaction products. Thus, fuel cells generate electricity as long as the reactants are supplied and the reaction products removed. In contrast to batteries, they do not need to be refilled after complete discharge (exhaustion of the reactants). A variety of fuel cells exist, which differ in their constitutive materials, temperature range of use, electrolyte nature or electrochemical reactions. The technologically more-advanced fuel cell is the PEMFC, in which the hydrogen oxidation reaction (HOR) and the oxygen reduction reaction (ORR) take place at the anode and cathode, respectively. The membrane electrode assembly (MEA) is the core component of the PEM fuel cell. It is an assembled stack of the proton exchange membrane, both anode and cathode catalyst layers and gas diffusion layers as shown on the scheme Figure 1.9.
Figure 1.9: Components of a PEMFC single cell: schematic (left); scanning electron microscopy (SEM) image of the MEA (middle) and transmission electron microscopy (TEM) images of a commercial Pt/C catalyst (right).
At the anode, the HOR produces protons and electrons according to: H2 → 2H+ + 2e− H0+/H2 = 0 V vs. RHE (1.3)
Where RHE is the Reversible Hydrogen Electrode. Those protons are forced to travel through the proton exchange membrane while electrons must take the external circuit to the cathode. At the cathode, protons recombine with electrons where oxygen is reduced according to: O + 4H++ 4e−→ 2H 2 O 0 /H O = 1.23 V vs. RHE (1.4)
Finally, these exothermic reactions produce water and heat according to the overall reaction:
∆ 0 = 1.23 V
2H2+ O2 → 2H2O (1.5)
∆r 0 = −237 kJ molH−12
Thermodynamically, a single cell voltage is 1.23 V. Nevertheless, several factors can induce cell voltage drops: Activation polarization: Mainly due to slow ORR kinetics and mixed potential caused by reactant permeation through the PEM.
Ohmic Polarization: Due to PEM and other components resistivity.
Concentration polarization: Reactants starvation at electrodes occurs for high current densities. These factors are cumulative and affect the cell voltage on the global current range. However, it is possible to associate a region of the polarization curve to its dominant overpotential cause (note that others are also present) as shown Figure 1.10.
Usually, in first approximation, those overpotentials are simply added to obtain the cell voltage for an operating current I: ( ) = =0 – ηORR( ) – ηHOR( ) – ηConc (I) – Ω (1.6)
Where ηi is the overpotential associated to reaction i and Ω the global electric resistance.
The Proton Exchange Membrane
The proton exchange membrane is the electrolyte of the PEM fuel cell. The three roles of the polymeric membrane in the PEM fuel cells are to:
Provide an ionic path for protons to travel from the anode to the cathode, Separate the reactant gases, Be an electronic insulator.
In general, materials used in synthesis of the polymer electrolyte membranes are classified into three vast groups according to the literature 17: perfluorinated ionomers (or partially perfluorinated), non-fluorinated hydrocarbons (including aliphatic or aromatic structures), and acid–base complexes. The most commonly used Nafion® membranes (Figure 1.11) developed by DuPont belong to the perfluorosulfonic acid (PFSA) membrane group.
Nafion® owes its mechanical integrity to the polytetrafluoethylene (PTFE) film structure and its ion exchange capacity from the perfluorinated side chain with terminal sulfonic acid groups. It can be underlined that the cost of such membrane technology (from $400 to $750 /m²) is currently one of the barriers for PEMFC development. For information, many promising researches based on radiation-grafted polymers 18,19 have been done recently to find alternative solutions to Nafion®. However, the polymer electrolyte membrane is currently still one of the components the most susceptible for failures. Three mechanisms can explain the membrane ageing 18 : Unzipping of polymer chains by radical attack. (Hydroxyl •OH or peroxy •OOH radicals) 19, 20.
Mechanical degradation: crack and pinhole formation induced by temperature and humidity fluctuations 21.
Thermal decomposition of the polymer.
A schematic of degradation mechanisms occurring into a PEMFC membrane has been proposed by de Bruijn et al. 22 and is shown on Figure 1.12:
Figure 1.12: Degradation conditions, mechanisms and effects for perfluorinated membranes. Adapted from Ref. 22
Many authors reported presence of fluorine ions, sulphate ions, and low-molecular weight perfluorosulfonic acid in drain water during PEMFC operation (23, 24, 25and 26) which is believed to be an excellent indicator of membrane chemical degradation. Combined with studies of exhausted gas at cathode outlet by mass spectrometry where the formation of HF, CO2, SOx and H2O2 under open circuit potential (OCV) durability tests has been shown 27, a chemical degradation mechanism could be proposed. In this mechanism, presence of oxygen at the anode side of the fuel cell because of cross-over, start/stop or air bleeding 28, leads, by reaction with hydrogen, to a production of H2O2 which can decompose to form the •OH or •OOH radicals. Even if they are not supposed to exist in currently chemically stabilized polymers according to Figure 1.11, polymer end groups with residual H-containing terminal bonds are believed to be formed during the polymer manufacturing process and may be present in the polymer in small quantities such as – CF2X where X = COOH for example 29. These weak end groups are privileged sites for radical attacks, inducing 3 steps:
Abstraction of hydrogen from an acid end group (step 1):
R−CF2COOH+•OH→R−CF2•+CO2+ H2O (1.7)
Perfluorocarbon radical reaction (step 2):
R−CF2•+•OH→R−CF2OH→R−COF+ HF (1.8)
Hydrolysis of the acid fluoride (step 3):
R−COF+H2O→R−COOH+HF (1.9)
At the end of step 3, weak H-containing terminal bounds are regenerated. This mechanism consequently ‘unzips’ a complete PFSA unit into HF, CO2 and other products. Besides, the reactant gas cross-over is believed to be a key factor in membrane degradation, but many other parameters can contribute to membrane ageing, particularly products from catalysts degradation as described in the following part.
The Catalyst Layers
Currently in most PEM fuel cell systems, the most common catalysts used in both the anode and cathode are platinum (or platinum alloy) nanoparticles supported onto high surface carbon. The use of a precious metal for innovative technologies with wide applications is not without raising the thorny issue of Pt resources. The DoE has established in 2017 technical goals for reduction of the total Pt group metal (PGM) loading (metal mass per unit of surface electrode) to 0.125 mg cm-2, targeting less than 10 g of precious metal per car by the year 2020 30. Strategies to reduce Pt loading in PEMFC catalysts will be further developed in a dedicated section.
Anode Catalyst.
Pure 2:The electrooxidation of hydrogen on platinum is well known to occur in two steps:
Dissociative adsorption of 2 to form (Heyrovsky and/or Tafel mechanisms):
H2 + Pt ↔ Pt-Hads + H+ + e− Heyrovsky (1.10)
H2 + 2Pt ↔ Pt-Hads +Pt-Hads Tafel (1.11)
Electrooxidation ofinto +(Volmer mechanism):
Pt-Hads ↔ Pt + H+ + e− Volmer (1.12)
This reaction is extremely fast, and is theoretically not a source of cell performance limitation (except in case of fuel starvation). But depending on the nature of the hydrogen, presence of pollutant (mainly CO) can strongly reduce anode reaction charges transfer as explained below.
CO poisoning:
The elimination of carbon monoxide is a current challenge in production of H2 from decarbonized fossil fuels 15. CO is very well known to decrease the performance of PEMFCs as it reduces the anode catalyst active surface area due to its strong adsorption onto the catalytic sites 31 depreciating the HOR kinetics, according to 32: CO + Pt = Pt-COads (1.13)
This Pt-COads bond is much stronger than any Pt-H bond, nevertheless this adsorbed COads can be electrooxidized at higher potential (0.6-0.9 V vs. RHE) according to the ‘reactant pair’ mechanism 33: Pt-CO + H O ↔ Pt + CO + 2H+ + 2e− (1.14) ads 2 2
This mechanism needs the formation of oxygenated species from the water molecule dissociation on Pt. Then, these oxygenated species oxidize the adsorbed COads. This poisoning issue is also occurring for Direct Methanol Fuel Cell (DMFC) where anodic methanol oxidation may lead to CO formation.
Pt-Ru anode catalyst:
It has been shown (34–37) that the use of alloyed or binary Pt-Ru as a catalyst at the anode strongly improves the PEMFC performance in presence of CO in the fuel. The enhancement of activity of Pt-Ru catalyst compared to Pt alone has been attributed to both:
A bifunctional effect 38,39: where the second (less noble) metal (here Ru) can generate OHads species from H2O at lower potential than Pt. The OHads species oxidize adsorbed COads on neighboring Pt catalytic sites (Equations (1.15) and (1.16)):
Ru + H2O ↔ Ru-OHads + H+ + e− (1.15)
Pt-CO + Ru-OH ads → Pt + Ru + CO + H+ + e− (1.16)
ads 2 A ligand effect (electronic interaction between Pt and Ru): where the presence of Ru reduces the strength of adsorption of CO on Pt, and so, facilitates the CO electrooxidation into CO2 34,40. This effect will be further described in a following section.
Naturally, this catalyst is preferentially chosen for PEMFC system working with hydrogen from hydrocarbons based fuel and in DMFC devices.
Cathode Catalyst
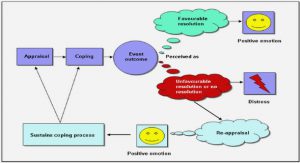
At the cathode, the ORR (a relatively complex and slow reaction compared to HOR) occurs. Its low kinetics is mainly responsible for PEMFC overall performance limitation and remains since many years a challenging problem in electrocatalysis 41. Several works in literature 41,42 highlighted two parallel mechanisms for oxygen reduction in acid environment as shown in Figure 1.13 and Equations (1.17)-(1.20): 4 electrons mechanism ‘direct pathway’: O2+ 4H++ 4 e− → 2H2O E0 = 1.23 V vs. RHE (1.17) O2 /H2O 2 electrons mechanism ‘peroxide or indirect pathway’ leading to 2 2 formation: O2+ 2H++ 2 e− → H2O2,ads EO0 /H O 2 = 0.67 V vs. RHE (1.18)
Followed by either H2O2,ads+ 2H++ 2e−→ 2H2O EH0 O 2 /H O = 1.77 V vs. RHE (1.19) Or 2H2O2 → 2H2O + O2 (1.20)
Based on rotating ring disk electrode experiments, several authors reported that the direct pathway on Pt is dominant, according to the small amount of H2O2 detected at the ring 41,42,44. However according to the theoretical model developed by Nørskov and co-workers, the direct pathway on Pt is itself composed of several steps 45: O + H+ + e− + Pt → Pt-OOH ads step 1 (1.21)
Pt-OOH ads + H+ + e− → Pt-O + H O step 2 (1.22)
ads 2
Pt-O + H+ + e− → Pt-OH ads step 3 (1.23)
ads
Pt-OHads + H+ + e− → H2O + Pt step 4 (1.24)
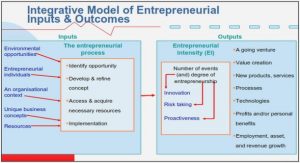
Changes in free energy compared to the final state for the different reaction steps (in the ‘direct pathway’) based on Density Functional Theory (DFT) calculations (from Rossmeisl et al. 46) give further insights on the origin of the rather low ORR kinetics on Pt. One can observe Figure 1.14, that if the free energy for the overall reaction is globally diminished, there are two reaction steps (steps 1 and 4) where the change in free energy is positive. These two steps are limiting the reaction kinetics, and the one with the higher positive shift appears to be the decisive step. In case of Pt, the most positive ∆G is associated to step 4, meaning that OHads intermediates are strongly bound to the surface, block the surface and inhibit the reaction kinetics.
Figure 1.14: Free energy diagram for the 4 ORR ‘direct pathway’ steps at 0.9 V vs. RHE on Pt(111). The overall reaction decreases in free energy, but 1st and 4th steps show positive free energy changes. From Ref. 46.
Many efforts have been dedicated to tailor the structure and the atomic composition of the surface and near-surface layers of Pt-based catalysts to decrease the binding energy of Pt-OHads intermediates, and a further detailed state-of-the-art on PEMFC cathode catalysts is proposed in the following chapter.
Carbon Support:
Carbon has been adopted for many years as widespread support for electrocatalyst because it’s high electrical conductivity, low cost and relatively good chemical stability. Some of the key properties of high-surface area carbon (HSAC) supports commonly used in PEMFCs are referenced in Table 1.3 47.
It may be seen that an ‘ideal’ electrocatalyst support must:
Have a high specific surface area (m2g−1) to efficiently spread the metallic nanoparticles and avoid their agglomeration,
Be chemically stable in the operating conditions of PEMFC anode and cathode, Have a high electronic conductivity.
Be porous enough to allow reactant access to catalytic sites and products evacuation.
Because the cathode operates at the highest potential in a fuel cell (from 0.6 to 1.2 V vs. RHE), it is subject to severe corrosion phenomena. The electrochemical corrosion of the HSAC support is a major contributor to the catalyst degradation. As carbon is corroded, noble metal nanoparticles are detached or aggregate to form larger particles, which cause a loss of electrochemically active surface area (ECSA) and of catalyst mass activity. Note also that mild corrosion of the HSAC leads to changes in surface hydrophobicity that can cause gas transport limitations 47. In the harsh operating conditions of a PEMFC (pH <1, 50-90°C) the following oxidation mechanism has been proposed: 48
Table of contents :
1 GENERAL INTRODUCTION
1.1 ENERGETIC CONTEXT
1.1.1 Global Warming, is There Time for Scepticism?
1.1.2 The Energetic Transition
1.2 PEMFC OVERVIEW
1.2.1 Principle
1.2.2 The Proton Exchange Membrane
1.2.3 The Catalyst Layers.
2 STATE-OF-THE-ART ON ORR ELECTROCATALYSIS
2.1 THE D-BAND THEORY FOR THE ORR
2.1.1 Investigations on Extended Surfaces
2.1.2 From Single Crystals to Nanoparticles
2.2 RECENT ADVANCES IN PEMFC CATHODE ELECTROCATALYSTS
2.2.1 Shape-Controlled PtM Nanoparticles
2.2.2 Pt-Based Core-Shell Nanoparticles
2.2.3 Hollow PtM Nanoparticles
2.3 PHD THESIS OUTLINE
3 HOLLOW PTNI/C NANOPARTICLES AS ELECTROCATALYST FOR THE ORR
3.1 INTRODUCTION
3.2 UNVEILING THE SYNTHESIS OF HOLLOW PTNI/C NANOPARTICLES
3.2.1 Methodology
3.2.2 Atomic-Scale Morphological Changes Occurring During the Synthesis of Hollow PtNi/C NPs
3.2.3 Structural Changes Occurring During the Synthesis of Hollow PtNi/C Nanoparticles
3.3 ELECTROCATALYTIC PERFORMANCE OF THE HOLLOW PTNI/C CATALYST
3.3.1 Something More than Strain and Ligand Effects
3.3.2 Structure-ORR Activity Causal Relationship
3.4 CONCLUSION
4 BEYOND ALLOYING EFFECTS: MICROSTRAIN-INDUCED ENHANCEMENT OF ELECTROCATALYTIC PROPERTIES ON VARIOUS PTNI/C NANOSTRUCTURES
4.1 INTRODUCTION
4.2 VARIOUS NANOCATALYSTS FROM PTNI NANO-BRICKS
4.2.1 Synthesis and Characterization of PtNi/C Nanoparticles
4.2.2 Structure and Chemical Composition of the Different Electrocatalysts
4.3 MICROSTRAIN AND ELECTROCATALYSIS
4.3.1 COads Stripping – ORR Catalytic Activity Relationships
4.3.2 DFT and Extension to Methanol and Ethanol Oxidation Reactions
4.4 CONCLUSION
5 FROM ‘PERFECT’ TO ‘DEFECTIVE’ PTNI ELECTROCATALYSTS: A MATTER OF TIME?
5.1 INTRODUCTION
5.2 PTNI MATERIALS SAMPLING FROM THE ORR ELECTROCATALYSIS LANDSCAPE 116
5.2.1 PtNi Materials Introduction and Synthesis
5.2.2 Materials Characterization
5.3 TOWARD UNPRECEDENTED ELECTROCATALYTIC TRENDS
5.3.1 From Microstrain to Surface Distortion
5.3.2 Facing the COads Stripping Puzzle
5.3.3 Apprehending ORR Catalysis Differently
5.4 INITIAL TRENDS VS. (ELECTRO)CHEMICAL AGEING.
5.4.1 Acidic Treatment
5.4.2 Accelerated Stress Tests
5.5 CONCLUSIONS
6 GENERAL CONCLUSION AND OUTLOOK
APPENDIX 1: MATERIALS AND METHODS
MATERIALS SYNTHESIS
XRD MEASUREMENTS
ELECTROCHEMICAL MEASUREMENTS
Characterizations
Accelerated Stress Tests
ELECTRON MICROSCOPY
OTHER CHARACTERIZATION TECHNIQUE
APPENDIX 2: MODELLING ALLOYING AND DISORDER
REFERENCES