Get Complete Project Material File(s) Now! »
HISTORY OF FATIGUE INVESTIGATION AND CLASSICAL CONCEPTS
The earliest observers of human performance must have already noticed a progressive decline of performance in intensely-used muscles, although this did not become part of scientific discourse until Berzelius described high levels of lactic acid in the muscles of an exhausted stag in 180712,13. Needham gives a detailed account of the history of muscle research in his Machina Carnis 14 tracing the aetiology of fatigue from Berzelius over Mosso’s 1904 book La fatica15, creating fatigue symptoms using electrical stimulation and the seminal paper of Hill and Kupalov16, indicating that muscular performance decrease was directly linked with lactic acid accumulation and dispersion16. These early studies gave rise to the notion that fatigue is a mainly peripheral phenomenon and strongly linked to lactic acid concentration. This idea was first challenged by Eberstein and Sandrow17, who perfused fatigued muscle fibres with caffeine and observed an acceleration of force production recovery, suggesting that fatigue may instead be linked to a failure of the excitation-contraction (E-C) coupling. Soon after, Bergström et al.18 perfected a muscle biopsy technique, which allowed the extraction of muscle samples for biochemical analysis at specific time points during exercise. This facilitated more detailed analysis of the biochemical changes involved in the fatigue process and in turn led to research identifying the causes of fatigue as linked to glycogen depletion19 and phosphate metabolite and H+ accumulation20. These muscle-centric changes are collectively known as peripheral causes of fatigue, as they are locally restricted changes in the force producing units. Investigations of these peripheral changes are often based on electrical stimulation that circumvents possible confounders in the activation process. When the development of fatigue in an integrated physiological system is to be described, the activation process from the supraspinal level through the central nervous system (CNS) needs to be taken into account. Changes in α motor neuron drive may precede and modify peripheral fatigue manifestations and can account for an important part of the fatigue process. While this was in some form evident to the pioneers of fatigue research15,21, direct identification and quantification of changes in the motor drive output that is finally effectuated at the level of the motor end plates, let alone at other points in the activation cascade, has proven to be technically challenging. Until appropriate methods were developed (notably fine-wire electromyography22), studies of changes in motor output were rather empirical and employed indirect markers such as hypnosis, prior mental conditioning and using conditioning cues23. This geographical and temporal dissociation of the phenomena has historically led to a distinctly dualistic perspective on fatigue, dividing processes into peripheral and central components. More recently, models have been proposed that re-unify the components and focus on the (afference and feed-forward based) interaction between central and peripheral components of fatigue24–29.
FACTORS INFLUENCING CALCIUM RELEASE AND CALCIUM RE-UPTAKE
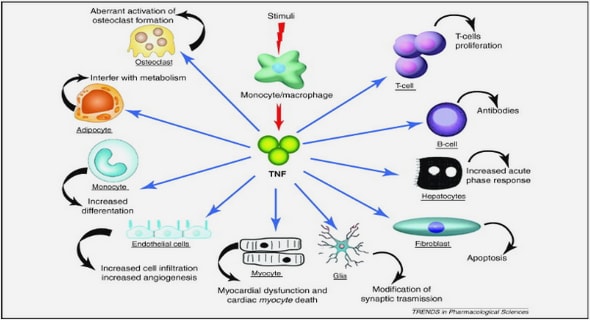
Once the AP has franchised the T-system, the next steps in the cascade leading to the release of Ca2+ from the SR are the activation of the voltage-sensitive DHPRs and the subsequent ryanodine receptor reaction. This sensitive part of the E-C coupling is termed the triad junction due to its three principle actors; the SR, the T-system and the DHPR-RyR sensor complex. In contrast to the actual processes in the T-system itself, the release of Ca2+ into the myoplasm is technically relatively straightforward to measure and provides an interesting outcome measure. In short, the interaction between the DHPRs and the associated RyRs calcium-release channels is currently a focal point of fatigue research46 and seems more bi-directional and complex than first assumed (for a detailed review consult Bannister47). Investigations of the triad junction indicate that the rate of Ca2+ release is dependent on myriad factors including the concentration48,49 and sensitivity of DHPRs and RyRs50,51 and stimulation type (caffeine, AP, etc). Acutely, the Ca2+ rate of release is reduced by a decrease in amplitude of the arriving AP, increase of cytoplasmic ionic magnesium (Mg2+) levels52–54, decrease in myoplasmic ATP concentration55,56, and a decrease in SR Ca2+ levels57,58. Calcium reuptake into the SR is central to keeping the diffusion gradient stable. The reuptake rate is negatively affected by fatigue processes, namely by increased levels of inorganic phosphate and ADP and the associated decreases in ATP55,59–61. Inorganic phosphate can seep into the SR through the ionic chloride channels at high intracellular concentrations, leading to CaPi precipitation effectively lowering free Ca2+ concentration and blocking the calcium release channels49. This mechanism is also susceptible to oxidative stress and increased reactive oxygen species (ROS) such as hydrogen peroxide62. Especially in prolonged exercise where the mitochondria adopt a high rate of ATP rephosphorylation, the development of ROS is an important contributor to the development of fatigue. Not only do ROS increase the influx of Pi into the SR, but they are also directly associated with c-Jun N-terminal kinase (JNK) -mediated necrotic cell death via the Tumour Necrosis Factor (TNF) Receptor super family63–65. Furthermore, ROS are implicated in the mediation of the inflammatory response, being secreted by myokines to induce mononuclear cell apoptosis66. The lack of Ca2+ re-uptake into the SR has also been demonstrated to be central in prolonged endurance exercise, as Ca2+ content has been shown to increase to over 30% of resting level following runs over 20 km67,68. Increased Ca2+ concentration and associated membrane leakage trigger signalling cascades via calpain activation, which ultimately result in further cell necrosis and mononuclear cell invasion69.
ATP SUPPLY AND METABOLIC BY-PRODUCTS
Adenosin tri-phospate (ATP) is the muscle cell’s primary source of energy. An ATP depletion lead to a loss in force production, as the unlatching of myosin cross bridges (CB) becomes impeded and cycling becomes impossible. Additionally, a number of ATP or ADP to Pi ratio sensitive mechanisms that can hinder the E-C coupling exist. A decrease in the ATP to ADP ratio for instance leads to a down-regulation of the Ca2+ release at the RyR site, a reduction in CB cycling velocity, a mitigation of CB catching force, a reduction in Ca2+ sensitivity, a decrease in Ca2+ resorption rate and increased CaPi precipitation in the SR. A fall of the ATP to ADP ratio also leads to an increase in adenosine monophosphate (AMP), which gives a strong signal to the AMP-activated protein kinase (AMPK) to curb energy depletion and reactivate catabolic ATP synthesis pathways70. ATP production is principally dependent on ready supplies of ADP, Pi, PCr, substrates and oxygen among others. Potential limitations in the substrates required for ATP synthesis can lead to a shift in synthesis pathways leading to a less optimal ratio of ATP to waste products. ATP during prolonged effort is mainly synthesised in the mitochondria through the aerobic pathway, this being by far the most efficient ATP synthesis process71. Without going into too much detail, mitochondrial abundance is one of the potentially limiting factors. Type 1 muscle fibres (red muscle) tend to represent the primary fibre type in endurance runners and exhibit greater mitochondrial abundance. Further bottlenecks are oxygen and nutrient supply. Oxygen supply is dependent on oxygen uptake at the level of the lungs, cardiac output and diffusion capacity (capillarisation) at the muscle site. A number of different energy sources can be used at the nutrient level, the main sources in prolonged exercise being composed of carbohydrates and fat. Glycogen is stored in the muscle cells themselves and primarily in the liver, providing a fast and highly effective energy source. Triglycerides and fatty acids on the other hand are a less optimal energy source, as cross-membrane transport is energy driven, and fatty acids must undergo oxidation before they can enter the Krebs cycle72,73. Substrate utilisation is finely regulated using different messaging cascades, the most central and well-known being the AMPK-insulin cascade71. The aerobic ATP synthesis pathway is also considered the main source of radical oxygen species (ROS) which have been shown to incite signalling cascades leading to inflammation and protein degradation. As the muscle becomes fatigued, the ATP synthesis provided by the aerobic pathway alone is no longer sufficient to meet the cell’s energy demands and the anaerobic pathway is re-stimulated. By-products of the anaerobic pathway include lactate (Lac), Pi, and H+ ions, both of which have detrimental effects on the force generating capacities of the cell. As [H+] increases, the pH of the cell drops decreasing Ca2+ sensitivity and Ca2+ resorption in the SR. Increased [Lac] was for a long time considered the primary instigator of fatigue, yet more recent studies indicate that the concentrations reached during exercise (< 30 mMol) are far below the threshold for force depression (50 mMol), Ca2+ release inhibition and Ca2+ sensitivity depression74. As lactate diffuses through the cell membrane, a diffusion gradient is created draws cellular water (H2O) into the extracellular matrix. This leads to a drop in intracellular [H2O] and an associated inhibition of force production. In short exercise this may be negligible, yet in prolonged exercise global dehydration frequently onsets and the muscular drainage process is exacerbated. In summary, the metabolic component of fatigue can be seen as the driving mechanic of the process. The compounds stemming from the metabolisation and rephosphorylation of ATP, mainly H+, Mg2+, ROS, and Pi, result in the processes ultimately leading to contraction failure through various interrelated pathways75.
MYOSIN CROSS BRIDGE LATCHING AND CYCLING
As already touched upon in the preceding section, the final possible site of fatigue is within the latching and cycling of the myosin cross bridges (CB) themselves. The process can be differentiated into a number of phases starting with the Ca2+ induced shift of the troponin complex, MHC latching (weak phase), ATP hydrolysis and power stroke, ATP binding (strong phase) and MHC release, Ca2+ regulation and movement of the troponin complex. Fatigue has been found to slow CB cycling, reduce maximal CB latching force and reduce myofibrillar Ca2+ sensitivity therefore decreasing the number of open binding sites for a given amount of Ca2+ 8. Discretely, Ca2+ sensitivity is reduced by an increase in [Pi], [ROS]76 or [H+]. Increases in [Pi] have also been associated with reduction in latching force, which may lead to a “slipping” of the myosin heads, making each cycle less effective. Increases of [ADP] have been shown to negatively impact CB cycling velocity, although the precise mechanisms are not yet fully elucidated.
METHODS USED TO DETERMINE PERIPHERAL FATIGUE IN TRAIL RUNNING
During trail running, the determination of peripheral fatigue is difficult due to limited available non-invasive methodologies. In order to stabilise and maximise motor drive, techniques such as evoked contractions and twitch trains of different lengths are frequently employed. The main muscle groups investigated are the knee extensors and the plantar flexors, both of which have shown force depression post trail running in function of exercise duration77–81. Stimulation is generally applied either neutrally (PNS) or percutaneously (Estim) at various frequencies. Millet et al.79 for instance employed PNS at stimulation frequencies of 20 and 80 Hz after a 30 km trail run (E/D = 27) in the knee extensors. Different frequencies were use in stimulation to try and identify low frequency fatigue (LFF), which is associated with peripheral alterations. They observed a depression in peak-to-peak amplitude of the electromyographic response to PNS (M-wave), and a decrease in mechanical twitch response amplitude and contraction time, although no changes were reported in the rate of twitch force development or relaxation. Both stimulation frequencies exhibited a similar decline in evoked force, leading to an unchanged frequency ratio79. Recently, LFF was observed for the first time in trail running77, yet this remains a novel occurrence and was recorded after a 161 km race, so might represent a special case. Similar methodologies have been used following mountain ultra marathons77 and prolonged trails81. Apart from these non-invasive strategies, it is feasible to recover tissue samples using muscle biopsies, although there is currently no published study examining tissue samples in trail runners. This would allow protein analysis, although the samples are probably prone to streaming due to the damaging nature of the exercise and there is an impact of the procedure itself82,83. Similar peripheral fatigue has also been extensively investigated in prolonged flat running84–86 of similar duration, although it has been suggested that the greater eccentric strain encountered during trail running evokes a specific type of peripheral fatigue77,87. Further investigation is warranted to determine whether or not trail running results in a different peripheral fatigue profile than other types of endurance exercise.
Table of contents :
Chapter 1 – What is it all about?
1.1. What is trail running?
1.1.1. Definition
1.2. The heritage of trail running
1.2.1. Evolution and trail running
1.2.2. Trail running before the 20th century
1.2.3. Trail running in modern society
1.2.4. Population statistics
1.3. Trail running as a model in science
1.3.1. Publication frequency
1.4. Conclusion
1.5. Chapter 1 bibliography
Chapter 2 – The aetiology of fatigue
2.1. Background
2.2. History of fatigue investigation and classical concepts
2.3. Peripheral fatigue
2.3.1. The excitation-contraction coupling
2.3.2. Potential fatigue processes affecting the propagation phase
2.3.3. Factors influencing calcium release and calcium re-uptake
2.3.4. ATP supply and metabolic by-products
2.3.5. Myosin cross-bridge latching and cycling
2.3.6. Methods used to determine peripheral fatigue in trail running
2.4. Central fatigue
2.4.1. Characteristics and investigation methods in central fatigue
2.4.2. The muscle wisdom hypothesis – spinal factors
2.4.3. Contributors to central fatigue – supraspinal factors
2.4.4. The “central fatigue hypothesis” – neurotransmission factors
2.4.5. Methods used to determine central fatigue in trail running
2.5. Fatigue in prolonged exercise
2.5.1. The “catastrophe theory”
2.5.2. Teleo-anticipatory models (CGM)
2.5.3. The flush model
2.6. Eccentric muscle damage
2.6.1. Muscle fatigue and muscle damage
2.6.1.1. Functional consequences of EIMD
2.6.2. Mechanisms of EIMD
2.6.2.1. Mechanical induction
2.6.2.2. Biochemical response to EIMD
2.6.2.3. Post-recovery adaptations to a single bout of EIMD
2.6.3. The repeated bout effect
2.7. Conclusion: Fatigue in trail running
2.8. Chapter 2 bibliography
Chapter 3 – Methods
3.1. General investigation design
3.1.1. Subjects
3.2. Metabolic responses
3.2.1. Maximal oxygen consumption and ventilatory threshold
3.2.2. Heart rate
3.2.3. Locomotion efficiency
3.2.4. Blood lactate concentration
3.2.5. Near infra-red spectroscopy
3.3. Psychological parameters
3.3.1. Rate of perceived exertion
3.4. Muscular function
3.4.1. Maximal voluntary isometric contraction
3.4.2. Percutaneous electric stimulation
3.4.3. Surface electromyography
3.4.4. Counter movement jump
3.4.5. Creatine kinase and lactate dehydrogenase
3.5. Thermal indexes
3.5.1. Core temperature
3.6. Chapter 3 bibliography
Chapter 4 – Descriptive Study
4.1. Introduction
4.2. Effects of a trail running competition on muscular performance and efficiency in welltrained young and master athletes
Chapter 5 – Reproducibility Study
5.1. Introduction
5.2. Reproducibility of performance and fatigue in trail running
Chapter 6 – Intervention Study 1
6.1. Introduction
6.2. The influence of wearing compression stockings on performance indicators and
physiological responses following a prolonged trail running exercise
Chapter 7 – Intervention Study 2
7.1. Introduction
7.2. Functional consequences of passive heating on exercise-induced muscle damage
Chapter 8 – Discussion
8.1. Synopsis of results
8.2. Integration into the existing literature framework
8.3. Critical assessment of methods
8.4. Implications for future research and outlook
8.5. Conclusion
8.6. Chapter 8 bibliography