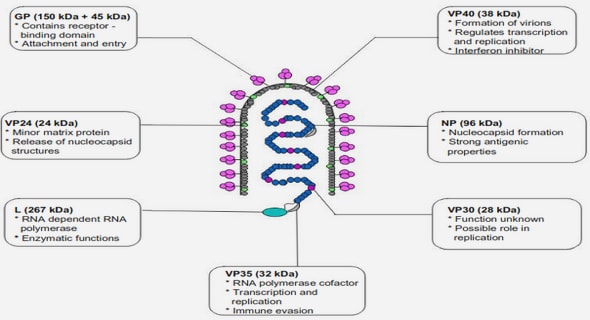
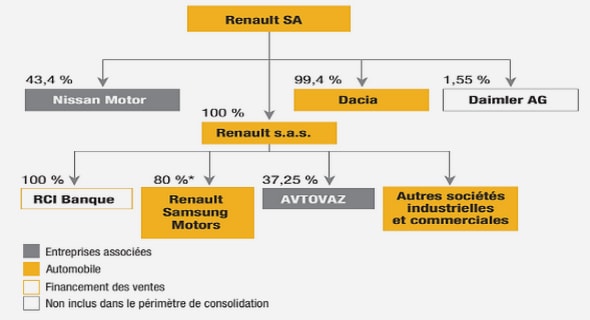
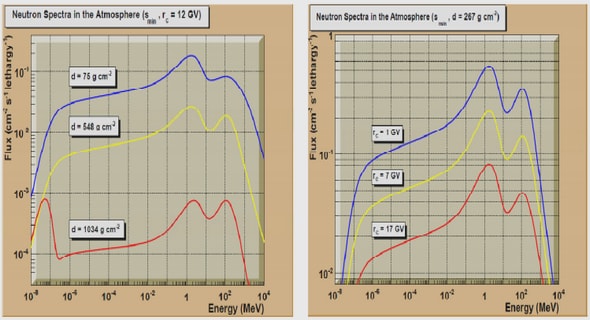
Get Complete Project Material File(s) Now! »
In – vitro culture of Actinidia species
Introduction
Culturing of plants in-vitro allows for the rapid propagation and maintenance of large numbers of plants under sterile conditions in a relatively small amount of space (Paprstein et al., 2008). In-vitro tissue culture of plant material is also used for germplasm conservation, exchange of vegetatively propagated crops, and elimination of viruses either through culturing alone, or enabling more efficient application of anti-viral treatments compared with application of treatments to whole plants (Gella & Errea, 1998; Knapp et al., 1995; Paprstein et al., 2008; Spiegel et al., 1993). The basis of virus elimination from plants using in-vitro tissue culture has been discussed in Chapter 1 (pages 26-28).
In order to evaluate the anti-viral effect of the chemotherapeutic and thermotherapeutic treatments studied during this project, a large supply of virus infected shoot tip cultures was needed. A model system (N. occidenatalis, described in Chapter 2) was available for initial treatment testing, but any promising treatments should then be further tested in Actinidia species. There are two distinct approaches to the production of virus infected plantlets in culture, either initiate cultures from virus infected field material, or infect healthy in-vitro cultured plantlets with virus.
In-vitro culture of Actinidia, such as A. deliciosa and A. chinensis, is generally considered reliable, with plants being easily micropropagated by axillary bud stimulation; the most viable propagation method for large-scale production of genetically uniform Actinidia species (Ferguson & Seal, 2008; Revilla et al., 1992; Rugini & Gutiérrez-Pesce, 2003). Meristem tip culture, while an important method in the elimination of viruses, would have been in-appropriate for initiating cultures, as the aim was to obtain virally infected Actinidia species cultures for the evaluation of the anti-viral effects of thermotherapy and chemotherapy. Furthermore, there have not been any reports of culture initiation from meristem tips, and meristem tip culture sometimes induces somaclonal variation in plants (Rani & Raina, 2000).
There is no literature on the mechanical inoculation of viruses into shoot tip cultures, but common methods of inoculation in whole plants include application of inoculum in conjunction with wounding the plant by rubbing with an abrasive (e.g. carborundum) or cutting with a blade, or by injecting the inoculum directly into the plant with a needle (Hull, 2002).
This chapter describes the in-vitro culture of Actinidia species from both virally infected and healthy shoot material, the mechanical inoculation of viruses into A. deliciosa, and the RT-PCR screening of presumed virus-free A. chinensis and A. deliciosa plantlets. Further descriptions of the plant material used can be found in Chapter 2, Table 2.1.
Methodology
In – vitro culture of Actinidi a species from virally infected material
The initiation of virally infected Actinidia cultures from infected cane material was carried out in order to obtain material for evaluating methods of virus elimination from Actinidia. M2 medium (Table 3.1), which has been routinely used for the tissue culture of A. chinensis and A. deliciosa at Plant and Food Research, Auckland, New Zealand (D. Cohen and T. Wang pers. com.) was used as the initiation medium for virus infected Actinidia species. Winter dormant canes of virus infected A. chinensis ‘Eexiang’ (AcVA and AcVB), A. chinensis ‘Hongyang’ (CLBV-a), A. chinensis ‘Lushanxiang’ (AcVA and AcVB), A. chinensis ‘Russell’ (AVX), and A. guilinensis (AMV) were chilled for four to six weeks at 4°C to break dormancy. Bud break was initiated by cutting canes into lengths with three or more nodes and then immersing the lower third of the cane in water and then leaving them under ambient temperature and lighting conditions in the laboratory.
Newly emerged shoots with at least four nodes were collected (after approximately four weeks) and then surface sterilised as described in Chapter 2, section 2.3.1. Newly emerged shoots were also collected from glasshouse-grown rooted canes of A. fortunatii (AMV and CMV), A. glaucophylla (AMV and CMV), and A. guilinensis (AMV). Surface sterilisation was carried out essentially as described in Chapter 2, section 2.3.1, except that the shoots were placed in cheese-cloth bags and left under running tap water for one hour prior to the application of sterilising solution. This was done since glasshouse-grown material was more likely to harbour microbial contaminants (Smith, 2000). Washing the material under running water first promotes germination of any fungal spores that may be present, which are then more easily killed by the sterilising solution (D. Cohen, Plant and Food Research, Auckland, New Zealand pers. comm.). Sterilised shoots were rinsed twice with sterile water in an Esco LHC-4A1 laminar flow cabinet (Esco Micro Pte Ltd, Singapore) and ~5mm nodal sections were cut from the stems.
The sections were then placed horizontally with the nodes facing upwards on M2 medium (refer to Table 3.1) in sterile vented 90x15mm plastic petri dishes (Labserv, Auckland, New Zealand). The petri dishes were sealed around the edges with cling film, and the explants were incubated at 25±1°C under a 16 hour photoperiod with cool white fluorescent light (~30µmol m-2 s-1). Any explants that displayed browning were transferred immediately to fresh M2 medium, as described by Preece and Compton (1991) and Torres (1989), to protect the nodal sections from the detrimental effects of phenolic exudation and oxidative damage.
Shoots (~5mm) that developed from the primary explants were sub-cultured onto fresh M2 medium in sterile 98mm round plastic tissue culture tubs (Intervet/Schering-Plough Animal Health, Boxmeer, The Netherlands) in order to further develop into plantlets. After this point, any shoots that were initiated were sub-cultured every month and incubated at 25±1°C under a 16 hour photoperiod with cool white fluorescent light (~30µmol m-2 s-1).
The A. chinensis ‘Hongyang’ shoots were cultured first on M2 media for one month, with movement of shoots displaying browning to fresh M2 medium. This was followed by sub-culturing onto M2 media supplemented with 100mg/L ascorbic acid (Sigma-Aldrich Inc., St. Louis, MO, USA) for one month, followed by one month on M2 media with 150mg/L citric acid (Serva, Heidelberg, Germany), followed by one month on M2 media with 100mg/L activated charcoal (Sigma-Aldrich Inc., St. Louis, MO, USA). These three substances are commonly used to reduce phenolic oxidation (Beyl, 2000; Bhatia & Ashwath, 2008; García-Gonzáles et al., 2010; Kärkönen et al., 1999; Noshad et al., 2009; Pan & Staden, 1998; Pierik, 1987; Shekhawat et al., 1993; Thomas, 2008).
Fungal contamination of A. chinensis ‘Hongyang’ explants was treated with Plant Preservative Mixture™ (PPM™), as per the manufacturer’s instructions (Plant Cell Technology Inc., Washington, DC, USA). Explants were cleaned with a soft tooth-brush under a stream of tap water, followed by continuous stirring on an Orbit™ 1000 platform shaker (Labnet International Inc., Edison, NJ, USA) at 100rpm in a 50% PPM™ solution (diluted with sterile water) for 15 minutes. Explants were then cultured without rinsing onto fresh M2 medium containing 0.2% PPM™ for one month. This treatment was also carried out on cultures of A. guilinensis displaying fungal contamination.
A. guilinensis and A. glaucophylla were sub-cultured on a monthly basis for two months on M2 media. Plantlets that were un-responsive (e.g. did not elongate) to the M2 media were cultured onto M5 media followed by M6 media and then M7 media for one month each (refer to Table 3.1). These media (M5-M7) were made with half strength Murashige & Skoog (MS) (Murashige & Skoog, 1962) basal salts and vitamins (Duchefa, Haarlem, The Netherlands). Although MS medium has been used effectively for the invitro culture of many plant species including some Actinidia, the level of salts can be too high or even toxic for some plants (Bairu et al., 2009b; Bhojwani & Razdan, 1996). The M5-M7 media also contained different combinations of the plant growth regulators (PGRs) cytokinin and auxin (refer to Table 3.1).
In addition, cane material of A. chinensis ‘Eexiang’ (AcVA and AcVB), A. chinensis ‘Lushanxiang’ (AcVA and AcVB), A. chinensis ‘Russell’ (RMV), and A. chinensis ‘Hongyang’ (CLBV-a) was sent to J. Claridge (Hetherdale Hortlab Ltd, Katikati, New Zealand) for establishment of tissue cultures, as she has expertise in the in-vitro culture of Actinidia species. T. Wang (Plant and Food Research, Auckland, New Zealand), who is also an expert in tissue culture of Actinidia species, attempted to establish cultures of A. chinensis ‘Hongyang’ (CLBV-a), A. guilinensis (AMV and CMV) and A. glaucophylla (AMV and CMV) from shoots initiated as described above.
Mechanical virus inoculation of A. deliciosa plantlets in – vitro
It became apparent that establishing cultures of virally infected Actinidia species from cane material was difficult, and it was not certain that if cultures were established that the plantlets would also be infected. As an alternative approach extracts of virally infected N. occidentalis plantlets were mechanically inoculated on to healthy A. deliciosa ‘Hayward’ growing in tissue culture.
A. deliciosa plantlets to be inoculated were incubated without light for 24 hours prior to inoculation. Inoculum preparation and application was carried out in Esco LHC-4A1 laminar flow cabinet (Esco Micro Pte Ltd, Singapore). Inoculum was prepared by grinding 100mg of leaf tissue from AMV, ASGV, AVX, or CLBV-a infected N. occidentalis plantlets in 9.9mL of sterile 0.1M sodium phosphate buffer (3.1g NaH2PO4∙H2O and 10.9g Na2HPO4 (anhydrous) in 1000mL distilled H2O) containing 5% polyvinylpyrrolidone and 0.12% sodium sulphite pH7.5, with a sterile mortar and pestle.
Inoculum was applied to eight replicate A. deliciosa plantlets in three different ways:
1. virus inoculum (AMV and CLBV-a only) was combined with sterile 400 mesh carborundum powder and rubbed onto three newly expanded leaves, left for one minute and then rinsed off with sterile water
2. virus inoculum was loaded into a sterile insulin syringe with a 26 gauge half inch needle (Becton, Dickinson and Company, Rutherford, NJ, USA) and injected into fleshy parts of the shoots (e.g. junctions between stem and petioles)
3. plantlets were submerged in virus inoculum and the stem and petioles were cut with a sterile scalpel blade, plantlets were left submerged for one minute and then rinsed with sterile water
Plantlets were then cultured and maintained as described in Chapter 2, section 2.3.2 and tested periodically by ELISA as described in Chapter 2, section 2.4.1. After five months the plantlets were tested by RT-PCR as described in Chapter 2, section 2.4.2.
In – vitro culture technique verification
The difficulties experienced with the establishment of tissue cultures from virally infected Actinidia species raised the question of whether it was virus presence or the technique used that was impacting on culture establishment. In order to investigate this, fresh attempts to initiate healthy cultures of A. chinensis ‘Hort16A’ and A. deliciosa ‘Hayward’ were made, since the healthy cultures used in this project had thus far been provided as already established plantlet cultures (refer to Chapter 2, section 2.2).
Canes of A. chinensis ‘Hort16A’ and A. deliciosa ‘Hayward’, which were believed to be free of the viruses in this study, were obtained from Plant and Food Research, Te Puke, New Zealand. Cultures were initiated (refer to Figure 3.1) as described in section 3.2.1, except that approximately half of the nodal sections were cultured on M3 media instead of M2 media, and any shoots that developed were sub-cultured on M4 media for one month (refer to Table 3.1). This was done as a comparison, because the PGRs and sucrose levels used in the M3 and M4 media proved effective for Wu (2010) in the initiation of five genotypes of A. chinensis (‘Hort16A’and four selections from the Plant and Food Research red-fleshed kiwifruit breeding program). After initiation all plantlets were maintained on M2 media by sub-culturing every month with incubation at 25±1°C under a 16 hour photoperiod with cool white fluorescent light (~30µmol m-2 s-1).
In addition, CMV was inoculated in-vivo, onto shoots of A. chinensis ‘Hort16A’ and A. deliciosa ‘Hayward’ that developed on canes from Plant and Food Research, Te Puke, New Zealand. Inoculum was prepared as described in section 3.3.1. Inoculum was then combined with sterile 400 mesh carborundum powder and rubbed onto three newly expanded leaves of ten newly emerged shoots (at least four nodes long) for each of the two species, left for one minute, and then rinsed off with sterile water. Inoculated shoots were left on the cane under ambient light and temperature conditions. Two weeks after inoculation newly emerged leaves on inoculated shoots were screened by RT-PCR as described in Chapter 2, section 2.4.2. Shoots that tested positive for CMV were selected for initiation of plantlet cultures, which was carried out as described above. After initiation all plantlets were maintained on M2 media by sub-culturing every month with incubation at 25±1°C under a 16 hour photoperiod with cool white fluorescent light (~30µmol m-2 s-1). Established plantlets were screened two months after initiation for CMV by RT-PCR as described in Chapter 2, section 2.4.2.
RT-PCR screening of healthy Actinidi a species plantlet cultures
The virus-free status of the Actinidia species plantlet cultures provided by T. Wang (Plant and Food Research, Auckland, New Zealand) (refer to Chapter 2, section 2.2) and initiated from cane material (refer to section 3.2.3) was tested by screening this material for AcVA, AcVB, AMV, ASGV, AVX, CMV and CLBV-a by RT-PCR. RT-PCR was carried out as described in Chapter 2, section 2.4.2.
Results
In – vitro culture of Actinidi a species from virally infected material A. chinensis ‘Hongyang’:
Plantlets were initiated for A. chinensis ‘Hongyang’, however these plantlets eventually died due to browning (i.e. browning spreading from the cut end of the explant accompanied in some cases by browning of the medium) and later shoot-tip necrosis (i.e. browning and early abscission of the youngest leaves followed by browning of the shoot apex and eventually the whole plantlet). In addition, some plantlets were lost to fungal contamination as the explants were either deemed to be too infested for the PPM™ treatment outlined in section 3.2.1, and so were discarded, or the plantlets became brown and died after treatment.
A. chinensis ‘Eexiang’, ‘Lushanxiang’, and ‘Russell’: The A. chinensis cultivars of ‘Eexiang’ (AcVA and AcVB) and ‘Lushanxiang’ (AcVA and AcVB) failed to produce any shoots from the nodal sections, which died due to browning. The A. chinensis ‘Russell’ cultivar (AVX) did produce shoots; however these failed to develop enough for excision from the nodal section and (along with the nodal section) also turned brown and died.
A. fortunatii : Most of the A. fortunatii (AMV and CMV) cane material died before any shoots were produced and any nodal segments that were obtained and put into culture turned brown and died before any shoots developed. A. guilinensis and A. glaucophylla : Shoots were produced for A. guilinensis (AMV and CMV) and A. glaucophylla (AMV and CMV) but these failed to establish on any of the four different media trialed (M2, M5-M7, refer to Table 3.1). These shoots eventually died due to shoot-tip necrosis. Some A. guilinensis shoots were also lost to fungal contamination, with explants being either discarded or failing to survive the PPM™ treatment detailed in section 3.2.1.
In summary, none of the virus infected material was successfully cultured on M2 medium even though this had previously been routinely used for healthy A. chinensis ‘Hort16A’ and A. deliciosa ‘Hayward’. Attempts by J. Claridge and T. Wang to establish cultures from A. chinensis ‘Eexiang’ (AcVA and AcVB), A. chinensis ‘Lushanxiang’ (AcVA and AcVB), A. chinensis ‘Russell’ (RMV), A. chinensis ‘Hongyang’ (CLBV-a), A. guilinensis (AMV and CMV) and A. glaucophylla (AMV and CMV) was also unsuccessful (data not shown).
Mechanical virus inoculation of A. deliciosa plantlets in – vitro
New growth from all virus inoculated A. deliciosa ‘Hayward’ plantlets tested negative for the presence of AMV, ASGV, AVX and CLBV-a by ELISA. RTPCR screening five months after inoculation detected AVX in 4/16 plantlets and CLBV-a in 4/24 plantlets. Of the three methods of inoculation used (i.e. rubbing, injection, and cutting), cutting was the easiest to apply, and 6/8 AVX and CLBV-a positive samples were inoculated in this manner.
Direct sequencing (Macrogen, Seoul, South Korea) confirmed the identity of AVX and CLBV-a in these A. deliciosa ‘Hayward’ plantlets. More of the CLBV-a inoculated plantlets may have been infected, but faint banding and non-specific products made it difficult to determine this conclusively (refer to Figure 3.2). AVX and CLBV-a positive (along with suspected CLBVa positive) plantlets were maintained and propagated as described in Chapter 2, section 2.3.2. However, when the plantlets and their progeny were rescreened by RT-PCR as described in Chapter 2, section 2.4.2 five months later, all plantlets tested negative for virus presence.
Table of contents
Abstract
Acknowledgments
Table of contents
List of figures
List of tables
List of abbreviations
1- Introduction
2 – Materials and Methods
2.1 Introduction
2.2 Plant material
2.3 In – vitro plant tissue culture
2.4 Virus detection
3- In – vitro culture of Actinidia species
3.1 Introduction
3.2 Methodology
3.3 Results
3.4 Discussion
4- Effects of thermotherapy on Actinidia cultures and viruses
4.1 Introduction
4.2 Methodology
4.3 Results
4.4 Discussion
5- Effects of chemotherapeutic agents on Actinidia viruses
5.1 Introduction
5.2 Methodology
5.3 Results
5.4 Discussion
6- Molecular characterisation of AMV from Actinidia
6.1 Introduction
6.2 Methodology
6.3 Results
6.4 Discussion
7- Concluding discussion
References
GET THE COMPLETE PROJECT
Virus elimination from Actinidia germplasm