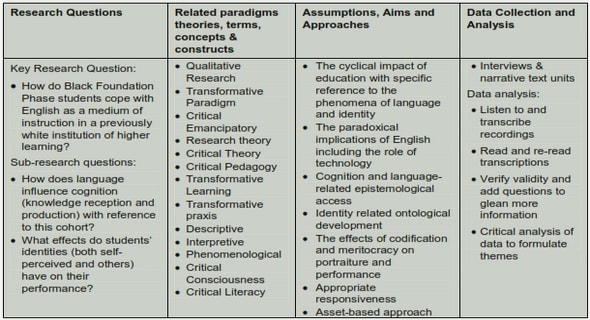
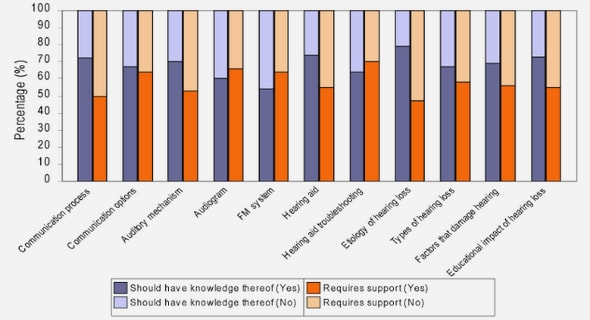
Get Complete Project Material File(s) Now! »
Gap closure and quality assessment
Initial contig ordering was performed by exploiting synteny with the preliminary genomic sequence of Ehrlichia chaffeensis, the closest relative of E. ruminantium for which genomic data were available at that time. The preliminary E. chaffeensis sequence was made available by The Institute for Genomic Research (www.tigr.org). The remaining gaps were filled by performing PCR amplification using all combinations of primers designed to anneal to the ends of all contigs. All primers were designed with annealing temperatures of 50-55oC. The PCR reactions contained 25 ng genomic DNA, PCR buffer, 0.25 μM of each primer, 0.2 mM dNTPs and 1 U TaKaRa Ex TaqÔ (TaKaRa Bio Inc.). Amplification was carried out under the following conditions: one cycle at 94°C (5 min), followed by 30 cycles at 94°C (10 s), 50°C (30 s) and 72°C (30 s), and a final extension at 72°C (7 min). When more than one amplicon was obtained, the PCR was repeated at a higher annealing temperature (53.5°C). PCR reactions which produced no amplification product were repeated at an annealing temperature of 48°C. Repeat regions, areas represented by single reads or clones, and regions of low quality were resequenced from PCR products generated from E. ruminantum (Welgevonden) genomic DNA. In total we designed and used 852 primers for gap closure (Appendix C1).
Particular attention was paid to ensuring the accuracy of the final sequence and all contigs were carefully examined to identify problems in the sequence. These problems included gaps in the sequence, weakly supported sequence, ambiguities in the sequence, and sequence on only one strand. The minimal criteria were established as either to obtain unambiguous sequence on both strands or, if sequence was available on only one strand, this had to be unambiguously confirmed on multiple clones, preferably from more than one library. The electropherogram data were used to edit sequences visually and, where discrepancies could not be resolved or a clear assignment made, the templates were resequenced or PCR amplicons were generated to obtain data of high quality. The same procedures were followed to check potential frameshifts, apparent chimeric sequences and areas containing repeats.
The integrity of the assembly was validated by comparing the positions of mapped genes and restriction sites to the physical map of De Villiers et al. (2000). A computed restriction map was created using the Staden package program Spin (Staden et al., 2000) and the recognition sites of the endonucleases KspI, RsrII and SmaI.
Selection of a gene set
The potential protein-coding genes were assigned by a combination of computer prediction and similarity searching. Three gene modelling programs, GeneMarkS (Besemer et al., 2001), Orpheus (Frishman et al., 1998) and Glimmer (Delcher et al., 1999), were used independently to predict potential protein coding sequences (CDSs). RBSfinder (http://www.tigr.org/software/) was used to assist with the location of start codons. When more than one potential start codon was identified, the first was arbitrarily chosen for annotation. The GC content, correlation scores and codon usage graphs from the Artemis sequence viewer and annotation tool (Rutherford et al., 2000) were also taken into consideration to select a gene set. Each CDS in the gene set was given a systematic identification number, starting with Erum0010.
In parallel, the entire genome sequence was used to search non-redundant protein databases (GenBank and Swiss-Prot/TrEMBL) with the BLASTx program (Altschul et al., 1997) to identify genes which were missed by the prediction algorithms. Transfer RNAs (tRNAs) were identified by tRNAscan-SE (Lowe & Eddy, 1997). If potential ORFs were partially or entirely overlapping, those showing similarity with known genes were chosen, and the longest one was selected unless the function of the shorter one was well supported in the databases.
CHAPTER 1: LITERATURE REVIEW
1.1. Heartwater
1.2. Genome sequencing
1.3. Aims of this study
CHAPTER 2: THE COMPLETION AND ANNOTATION OF THE GENOME SEQUENCE OF EHRLICHIA RUMINANTIUM (WELGEVONDEN)
2.1. Introduction
2.2. Materials and Methods
2.3. Results and Discussion
2.4. Conclusions
CHAPTER 3: METABOLIC RECONSTRUCTION AND COMPARATIVE GENOMIC ANALYSIS OF SPECIES WITHIN THE ORDER RICKETTSIALES
3.1. Introduction
3.2. Materials and Methods
3.3. Results and Discussion
3.4. Conclusions
CHAPTER 4: REPETITIVE DNA IN THE COMPLETE GENOME SEQUENCE OF EHRLICHIA RUMINANTIUM (WELGEVONDEN)
4.1. Introduction
4.2. Materials and Methods
4.3. Results and Discussion
4.4. Conclusions
CHAPTER 5: SELECTION OF POSSIBLE VACCINE CANDIDATES
5.1. Introduction
5.2. Materials and Methods
5.3. Results and Discussion
5.4. Conclusions
CHAPTER 6: CONCLUDING DISCUSSION
APPENDIX A: REFERENCES
APPENDIX B: MATERIALS, BUFFERS, MEDIA AND SOLUTIONS
APPENDIX C: PRIMERS
APPENDIX D: PROTEIN CLASSIFICATION SCHEME
APPENDIX E: E. RUMINANTIUM GENE LIST
APPENDIX F: WEB BASED TOOLS
APPENDIX G: PUBLICATIONS AND ETHICS