Get Complete Project Material File(s) Now! »
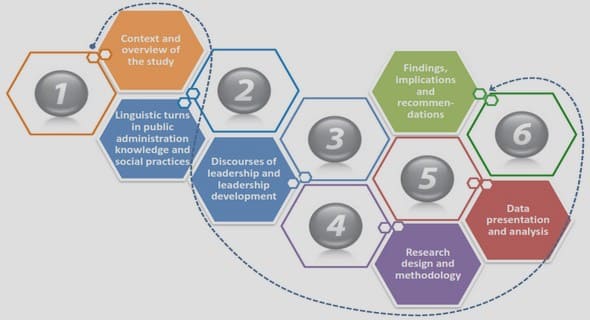
Chapter 9 Maturity Level 2
Introduction
In chapter 8, the KPAs applicable at all maturity levels were discussed. Essentially, they perform a facilitating function by supporting timely delivery of projects within budget and quality standard. As such, the soft skills management KPA helps in enhancing communication, building a team spirit, managing stress, etc. Given that a software project is under development, the strengths and weaknesses of software projects are taken into account. Similarly, the change management KPA and the integration management KPA is applied at all maturity levels as changes may occur at any stage during software development and all the KPAs have to be applied in an integrated manner rather than in isolation.
This chapter brings into perspective the KPAs that provide a core function of project management. Maturity level 2 of the ESPM3 is described as the basic project management level. It comprises three KPAs, namely:
Time management.
Cost management.
Quality management.
Software projects suffer from a lack of direct visible features regarding duration, progress and quality as compared to most traditional engineering projects (Sukhoo et al., 2005b). Prior to the delivery of a software product, the user may not know in advance whether or not the software will satisfy his/her requirements satisfactorily. Therefore, it is important through the use of appropriate tools and techniques to render visible the progress of software projects. All the three variables (time, cost and quality) are carefully controlled so as to improve the success rate of software projects. These variables have been depicted as the apex of a triangle, sometimes called the golden triangle of cost, time and quality (Gardiner and Stewart, 2000), as shown in figure 9.2:
Sometimes, project scope is used as a fourth variable as in the case of PMBOK®. In the case of the proposed model (ESPM3), project scope is taken care of in project quality.
This chapter elaborates on the three key process areas, the tools and techniques available to make an attempt to deliver software projects on time, within cost and as per initially set quality standard. Suggestions are also made with regards to the tools and techniques that Mauritian software organisations can consider.
Aim, objectives and structure of this chapter
In chapter 9, the maturity level 2 KPAs are described in greater detail than in chapter 7. The application of the Plan-Do-Check-Act cycle to the various KPAs is also explained.
The remainder of the chapter is structured as follows:
Section 9.2 – In this section, a conceptual representation of maturity level 2 is shown diagrammatically and explained.
Section 9.3 to 9.5 – All the KPAs are described in detail in these sections and the PDCA cycle as applied to these KPAs is made clear.
Section 9.6 – In this section, the evolution from maturity level 1 to level 2 is explained.
Section 9.7 – The conclusion aims at showing the importance of the various KPAs as discussed in previous sections.
KPAs at Maturity level 2
In addition to the KPAs specific for maturity level 2, the continuous process improvement group of KPAs provides a facilitating function to the core function KPAs. Soft skills management ensures that a conducive environment prevails during software development. Change management permits changes in requirements to be accepted with minimum disturbances to the schedule, cost and quality. Strengths associated with software projects are taken into consideration (for example, the use of evolutionary prototyping can clear up misunderstandings at an early stage during the software development process) and as far as possible, inherent weaknesses are mitigated (for example, invisible features like progress of the software development process can be rendered visible through the use of appropriate tools such as Gantt charts and the Critical Path Method). Environmental management, like responding to political and clients’ pressures, needs to be addressed at the same time when the KPAs responsible for the core functions (Time, Cost and Quality) are being considered. In addition, the integration management KPA ensures that all other KPAs are executed in a coherent manner thus allowing software development to proceed in a streamlined manner.
The KPAs at maturity level 2 (also discussed in chapter 7) is revisited below:
Maturity level 2, M2, is the union of all KPAs at level 1 (M1), all KPAs at the current level and the continuous process improvement group of KPAs (Mc). Given that M1 = Ø,
n
M2 = M1 U U KPA2j U Mc
j = 1
A total of 3 specific KPAs are identified at maturity level 2 and, therefore, n=3 in the above equation. Thus,
M2 = U KPA2j U Mc
j = 1
The KPAs are as follows:
KPA21 = Time management
KPA22 = Cost management
KPA23 = Quality management
The KPAs are developed till maturity at level 2 is reached as shown in figure 9.3.
This chapter focuses on the specific KPAs for maturity level 2, while chapter 8 provided an in-depth discussion on the continuous process improvement group of KPAs (Mc).
Time management
Time management refers to the processes involved in the delivery of software projects within a specified time period. Late delivery of software is a major source of conflict between software development organisations and clients with the result that a software contract may be cancelled and penalties claimed as damages. The matter may not be as simple as that for the clients since one software project may depend on the successful delivery of another one. It is, therefore, important for software developers to be committed to fulfil their contracts as agreed upon with their clients.
Time management involves the tracking of software projects as per the initial plan. Time management is mapped onto 4 process groups as per the ISO 9001:2000 standard. The development of the time management KPA is a requirement for an organisation to achieve maturity level 2. The KPA is developed progressively until it attains maturity at level 2.
It is plausible that a good starting point for time management is to produce a Work Breakdown Structure (WBS) or Product Breakdown Structure (PBS) and generate a list of activities. These activities can each be assigned a duration for completion. It must be noted that some activities may follow others, while other activities may occur concurrently. In ESPM3, time management can be carried out from the following inputs:
List of activities.
Constraints.
Resources.
Assumptions.
Historical data.
In ESPM3, the following outputs are generated:
A project schedule.
Documentation.
The transformation of the inputs to the outputs is shown in figure 9.4:
Time management is mapped onto four process groups, namely plan, do, check and act as per figure 9.5.
Plan: Time management
Time is a very important resource in project management. According to Peter Drucker (cited in Gray and Larson, 2000), time is the scarcest resource. Unless it is managed nothing else can be managed. The planning stage sets the target for the overall project as well as for the different tasks to be undertaken. The use of a time schedule renders the duration of the project and its activities visible. Planning the project’s duration in ESPM3 can be effected by several tools and techniques like:
Time estimation by analogy.
Expert judgement.
Simulation.
Gantt chart.
Network diagram.
The use of Gantt charts is quite popular in Mauritius. According to a survey carried out by Sukhoo et al. (2004b), around 70% of software development organisations make use of Gantt charts to generate their project schedules. Network diagrams are not as popular in Mauritius. According to the survey carried out by Sukhoo et al. (2004b), around 20% of software development organisations made use of network diagrams to plan the progress of software projects. However, network diagrams may provide useful information and can be considered together with Gantt charts along with the proposed ESPM3.
Do: Time management
The execution of a project is carried out according to the plan set out in the previous process group. As the activities are carried out, the progress is displayed so that the actual progress can be compared with the planned one. The Gantt chart provides useful information on the progress of activities. For example in figure 9.6, the progress of the actual analysis activity of a hypothetical project represents 50% completion (represented by the thick line in the horizontal column), while the other dependent activities have not yet started.
Check: Time management
Measurements are taken regarding the progress of the project. The actual progress is compared against the planned progress. An actual progress exceeding the time that was planned indicates a schedule overrun. This can cause delays in other activities, thus, resulting in the project running behind schedule.
However, a premature completion of an activity is welcomed as it represents a saving in cost, provided other activities are not delayed later.
Act: Time management
Any deviation from the plan as far as a delay in the completion of an activity is concerned is dealt with during the act process group. The reasons for the deviation need to be identified and action is taken to bring the project within schedule. Depending on the nature of the problem, the project manager may choose among a number of solutions like:
Adding more people on the project.
Extending the hours of work.
Outsourcing certain activities.
However, the real-life situation is even more complex. Adding more people to a project or on certain tasks may not always reduce the time factor as discussed by Sukhoo et al. (2004a). The project manager and software team members may deal with multiple project executions at the same time. In such a case, a master schedule needs to be prepared to provide a bigger picture of all projects being managed. The master schedule prepared in the form of a Gantt chart displays the various projects undertaken as well as the progress of each project. An example is shown in figure 9.7.
Cost management
Cost management refers to the process that ensures that a software product is delivered within the agreed cost. The cost may be an integral part of any software project management plan. Underestimation of the cost of software results in the loss of money on the part of the developer while overestimation results in a loss of the contract as the client may decide not to go ahead on the basis of a cost-benefit analysis or return on investment.
According to Schach (2002), two types of cost are associated with software development. The first type is called internal cost, which is the cost to the developers while the second one is called external cost and it is the cost to the clients. The external cost is the sum of the internal cost and a profit.
Cost management can begin with the identification of all inputs and the process to transform them into outputs. A Work Breakdown Structure (WBS) or Product Breakdown Structure (PBS) is used as input along with the resources required and historical information. In ESPM3, cost management can be carried out from the following inputs:
Work Breakdown Structure.
Project schedule.
Resources.
Historical data.
The inputs of the process generate outputs in the form of:
Cost estimation.
Documentation.
The transformation of the inputs to the outputs is shown in figure 9.8:
Cost management is mapped onto four process groups (the PDCA cycle), namely plan, do, check and act as shown in figure 9.9.
Plan: Cost management
Planning for cost is fundamental to the software development organisation so as not to cause an overrun leading to the loss of money. The cost of a software product is also linked to the duration of the project. If the project is stretched over a longer period, the actual cost may deviate from the planned one. The difficulty of software estimation lies in the hidden complexity and invisibility features inherent in software. It is only when the project progresses that a real cost is available, particularly when the complexity and invisibility features unfold. In ESPM3, planning the project’s cost can be effected by several tools and techniques like:
Analogy.
Expert judgement.
FFP (Files, flows and processes).
COCOMO (Constructive Cost Model).
Function point analysis.
Object point analysis.
Many organisations in Mauritius are using 4GL development platforms to build applications in Oracle, Microsoft Access, SQL Server, etc. (Sukhoo et al., 2004b). Object points analysis (Hughes and Cotterell, 2006; Parthasarathy, 2008) can provide a useful means to estimate the cost of software. This technique uses the number of screens, reports and 4GL components as well as complexity (measured in terms of simple complexity, medium complexity and high complexity) weightings. As many objects may be available within an organisation and do not require development, the number of object points is adjusted. For example, if 20% of objects are already available and a total of 840 object points have been computed, the adjusted number of object points (NOP) is given by (Hughes and Cotterell, 2006):
NOP = 840 * (100-20)/100 = 672
Furthermore, the number of person-months is computed by dividing a productivity rate (PROD) into the NOP. Past data is used to determine the PROD as shown in table 9.1.
Therefore, for a development environment where productivity is nominal, the estimated effort is 672/13 = 52 person-months. The cost of development is then calculated based on the rate per person-month for the organisation.
Do: Cost management
As the software development project progresses against its schedule, the different internal costs involved are recorded. A breakdown of the cost per module can be kept (see figure 9.10). These costs can be useful for later comparison purposes especially when a bottom-up estimation strategy has been applied. The cost involved may be tabulated using a software tool like Microsoft Project as shown in figure 9.10.
Check: Cost management
This process group allows the software developer or project manager to measure and compare the actual cost with the planned cost as the project proceeds. This is carried out for all activities to ensure that they are in line with the planned estimates.
Act: Cost management
Any deviation from the planned estimates must initiate an action on the part of the project manager to determine the reasons for any significant cost overrun. The use of appropriate software tools is recommended. For example, Microsoft Project or any other suitable software tool can show the progress of the activities along with their planned and actual costs. Therefore, the project manager can track cost at a glance with such software tools, provided the database is always kept up to date.
Quality management
By adapting the definition of Webster’s New World Dictionary, software quality can be defined as the degree of excellence of the software. The purpose of quality management is to ensure that the project will satisfy the needs for which it was undertaken (Schwalbe, 2004). According to Munro-Faure L., Munro-Faure M. and Bones (1994), the benefits that can accrue from the implementation of a successful quality management system are:
Improved customer satisfaction.
Elimination of errors and waste.
Reduced operating costs.
Increased motivation and commitment from employees.
Increased profitability and competitiveness.
Quality management can begin with the identification of all inputs and the processes to transform them into outputs. The following inputs to the quality management process are identified:
User requirements.
Quality standards.
Functional specifications.
Quality measurement criteria.
The processes generate outputs in the form of:
Quality management plan.
Quality management checklists.
Products conforming to standards and criteria.
The transformation of the inputs to the outputs is shown in figure 9.11:
Quality management is mapped onto four process groups (the PDCA cycle), namely plan, do, check and act as per figure 9.12.
In ESPM3, quality management is initiated by defining the following criteria:
Functionality.
Usability.
Reliability.
Performance.
Serviceability.
The implementation of a quality management system by the developer organisation may add other criteria to the above list.
Plan: Quality management
In ESPM3, a quality plan is prepared prior to the development of any software product. The desired quality targets need to be specified at this stage. The approach to be used, the implementation strategy and the major products have to be made clear at this stage. In the case of Mauritian software development organisations, ISO 9001:2000 certification is increasingly being sought after. Therefore, a quality management system to be implemented in the Mauritian context needs to focus on the requirements of ISO 9001:2000.
Do: Quality management
The plan is executed during the development of the software and associated documentation. The implementation of the plan has to be closely monitored. The use of checkpoints to determine quality at defined stages can provide useful measures.
Table of Contents
Part 1 – Introduction and Background
Chapter 1: Introduction and Background
1.1 Introduction
1.2 Background
1.3 Aim of the research
1.4 Problem Statement, objectives and deliverables
1.5 Research Approach
1.6 Thesis layout
1.7 Conclusion
Part 2 – Literature survey and status of Software Project Management
Chapter 2: Software project management principles and methodologies/models/standards
2.0 Chapter layout
2.1 Introduction
2.2 History of Project Management
2.3 Software project management principles
2.4 Available project management methodologies and models
2.5 Conclusion
Chapter 3: Approaches to project management in African countries
3.0 Chapter layout
3.1 Introduction
3.2 The state of IT and Software Project Management in the US and the UK
3.3 The state of IT and Software Project Management in African Developing Countries
3.4 Conclusion
Chapter 4: Software project management status in Mauritius
4.0 Chapter layout
4.1 Introduction
4.2 Survey of software project management tools, techniques and methodologies in Mauritius
4.3 Status of software project management in the public sector
4.4 Conclusion
Chapter 5: Current project management maturity models
5.0 Chapter layout
5.1 Introduction
5.2 Berkeley Project Management Process Maturity Model (PM)2
5.3 Microframe’s Self Assessment Tool
5.4 PRINCE 2 Maturity Model
5.5 Organizational Project Management Maturity Model (OPM3TM)
5.6 Kerzner’s Management Maturity Model
5.7 SEI’s Capability Maturity Model Integration (CMMI)
5.8 ISO/IEC Software Process Assessment
5.9 Conclusion
Chapter 6: Assessment of software project management maturity level in Mauritius
6.0 Chapter layout
6.1 Introduction
6.2 Assessment of software project management maturity in Mauritius
6.3 Conclusion
Part 3 – Proposed maturity models
Chapter 7: Overview of the Evolutionary Software Project Management Maturity Model
7.0 Chapter layout
7.1 Introduction
7.2 Motivation for a new software project management methodology
7.3 Unsuitability of PMBOK®, PRINCE 2® and CMMI for Mauritius
7.4 Survey/interview conducted in Mauritius
7.5 The Evolutionary Software Project Management Maturity Model (ESPM3)
7.6 Suitability of ESPM3 for Mauritius
7.7 Conclusion
Chapter 8: Continuous process improvement
8.0 Chapter layout
8.1 Introduction
8.2 Representation of continuous process improvement group of KPAs
8.3 Soft Skills Management
8.4 Change Management
8.5 Software specific focus
8.6 Environmental management
8.7 Integration management
8.8 Conclusion
Chapter 9: Maturity level 2
9.0 Chapter layout
9.1 Introduction
9.2 KPAs at Maturity level 2
9.3 Time management
9.4 Cost management
9.5 Quality management
9.6 The evolutionary process from maturity level 1 to maturity level 2
9.7 Conclusion
Chapter 10: Maturity level 3
10.0 Chapter layout
10.1 Introduction
10.2 KPAs at Maturity level 3
10.3 Human Resource Management
10.4 Risk Management
10.5 Contract management
10.6 The evolutionary process from maturity level 2 to maturity level 3
10.7 Conclusion
Chapter 11: Summary and Assessment of Evolutionary software project management maturity model
11.0 Chapter layout
11.1 Introduction
11.2 Conceptual representation of ESPM3
11.3 Continuous process improvement group
11.4 Maturity level 1
11.5 Maturity level 2
11.6 Maturity level 3
11.7 Assessment of maturity level in ESPM3
11.8 Conclusion
Chapter 12: Case Studies
12.0 Chapter layout
12.1 Introduction
12.2 Case study 1
12.3 Case study 2
12.4 Conclusion
Part 4 – Conclusion and Future Work
Chapter 13: Summary, Conclusion and Future Work
13.0 Chapter layout
13.1 Introduction
13.2 Contribution of this thesis
13.3 Analysis
13.4 Strengths of ESPM3
13.5 Weaknesses of ESPM3
13.6 Future Work
References
GET THE COMPLETE PROJECT