Get Complete Project Material File(s) Now! »
The mycobacteria
The Mycobacterium genus belongs to the Mycobacteriaceae family, the Actinomycetales order and the Actinobacteria phylum (Stackebrandt et al., 1997).
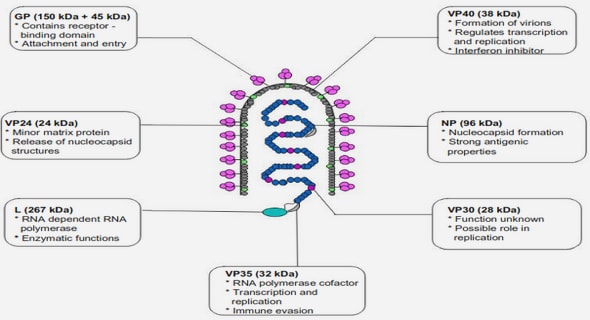
Mycobacterium species are non-motile, non-flagellated and non-spore forming Gram-positive aerobe. The bacilli are straight or slightly curved measuring 0.2 to 0.6μm in diameter and 1 to 10μm in length (Murray et al., 2003). One of the major characteristics of mycobacteria is their exceptionally rich lipid walls. These species harbor a unique, waxy layer composed of long chained mycolic acid, which gives high impermeability to the cell wall, making them naturally resistant to many antibiotics and helping them to escape immune clearance (McMurray, 1996). In addition, this gives them the ability of resisting to disinfectants, acids, and strong bases. From a diagnostic perspective, this lipid-rich cell wall enables the differentiation from most other bacteria by staining techniques, as mycobacteria resist decolorization with an acid-alcohol wash, hence their name of acid-fast bacilli (AFB) (Farver and Jagirdar, 2018). Another major characteristic of mycobacteria is their (relatively) slow growth when compared to other bacteria, as at least 5-7 days of culture are required to detect growth (Forbes et al., 2018).
Results from phylogenetic analysis support the taxonomic division of Mycobacterium species into slow-growers and fast-growers, requiring respectively, more or less than 7 days to grow on Lowenstein-Jensen (LJ) solid media (Gutierrez et al., 2009). Furthermore, mycobacterial sequence
comparisons of 16S rRNA sequences suggest the evolution of the slow-growing sub-group from a common fast-growing ancestor (Rogall et al., 1990).
Among the 170 or so slow- and rapid-growing Mycobacterium species described so far, the great majority were isolated from the environment. All but those included in the so-called Mycobacterium tuberculosis complex (MTBC) and M. canettii (also collectively called the TB bacilli) are designated as non-tuberculous mycobacteria (NTM) (Fedrizzi et al., 2017). While most fast-growing NTM are considered as harmless saprophytic organisms, a number of fast-growing NTM as M. abscessus, as well as slow-growing species as M. avium, M. marinum, M. xenopi, M. gordonae and M. kansasii can cause disease, mostly in immuno-compromised individuals (Figure 1). In contrast, the TB bacilli and the other human pathogens M. ulcerans and M. leprae are slow growers (Gutierrez et al., 2009).
M. tuberculosis genome, evolution and phylogeny
In addition to the principal agent of human TB M. tuberculosis, the MTBC comprise several additional members. M. bovis was the first animal-associated MTBC member discovered in 1896 and is primarily characterized as a pathogen of cattle. However, M. bovis has been isolated from a range of other mammalians and can also cause TB in humans (Smith et al., 2009). Other animal-associated MTBC members often appear to have a stricter host range and include (preferred host in brackets): M. microti (voles), M. pinnipedii (seals and sea lions), M. orygis (antelopes), M. mungi (banded mongooses), M. suricattae (meerkats), the dassie bacillus (hyrax), and the chimpanzee bacillus (Niemann et al., 2016; Gagneux, 2018).
The MTBC members all share identical 16S rRNA gene sequences, have more than 99% identity at the nucleotide level, and possess highly syntenic genomes. For these reasons, they are regarded as host- (human- or animal-) adapted variants (also termed ecotypes) rather than different species (Smith et al., 2009).
The first genome representative of the MTBC that was completely sequenced was from the type strain of M. tuberculosis H37Rv, in 1998 in collaboration between the Institut Pasteur in Paris, France, and the Sanger Institute in Hinxton, United Kingdom. This genome comprises 4411532 base pairs (bp), about 4000 genes and a G+C rich content (65.6%) (Cole et al., 1998). Among its other characteristics, it is rich in repetitive sequences, comprising in particular numerous genes encoding the so-called PE (Proline-Glutamate)/ PPE (Proline- Proline- Glutamate) protein families (see below) (Mathema et al., 2006).
With 4.34 Mbp, the genome of M. bovis is slightly smaller, reflecting deletions of some genome regions relatively to M. tuberculosis. The presence of such deletions (also termed regions of difference (RDs)) in the genomes of strains of M. bovis and other animal-adapted MTBC members argues against the zoonotic origin of TB. The characterization of the sequences and the distribution of these RDs among MTBC and M. canettii strains suggests that all MTBC strains derive by reductive evolution from a single clonal progenitor resembling more to M. tuberculosis or M. canettii (Brosch et al., 2002).
This scenario is further supported by results from comparative genomics of M. canettii strains, which correspond to exceptional TB clinical isolates from East Africa. These strains have slightly
larger genomes than M. tuberculosis, specific CRISPR-Cas systems, much greater genetic diversity, and multiple traces of horizontal DNA exchange contrasting with the strong clonality of M. tuberculosis (Supply et al., 2013; Boritsch et al., 2016). These data suggest that M. canettii represents evolutionarily early branching lineages of TB bacilli. In addition, mouse infection experiments showed that smooth tubercle bacilli (STB) strains are less persistent and virulent than M. tuberculosis. These results suggest that the MTBC ancestor emerged as a professional pathogen from an ancestral M. canettii-like pool of mycobacteria, putatively associated with an environmental reservoir in East Africa, by gain of persistence and virulence mechanisms (Supply et al., 2013). One such mechanism consists of a change in cellular morphotype due to recombination in the pks5 locus in M. tuberculosis, revealing a key step in pathoadaptation (Boritsch and Brosch, 2016).
Tuberculosis pathology and infection cycle
TB is predominantly a pulmonary disease but can also exhibit extra-pulmonary forms resulting from hematogenous dissemination and affecting e.g. pleura, lymph nodes, abdomen, genitourinary tract, skin, joints, and bones, or meninges (Lee, 2015). Symptoms vary by the site of infection, but the most common ones, associated with the dominant pulmonary forms, are a cough with bloody sputum, chest pains, weakness, weight loss, fever and night sweats (WHO, 2018e).
Infection usually starts with the inhalation of contaminated droplets, emitted into the air by a patient with pulmonary TB especially while coughing. After coughing or sneezing, the infectious particles can remain in the air for several hours and the minimal infectious dose can be very low, ranging from 1 to 10 bacilli (Russell et al., 2009). Tubercle bacilli can then be inhaled by contacts of TB patients and dragged by the mucosal surface to the lungs. In the case of TB caused by M. bovis, infection can occur via the digestive tract after the consumption of e.g. contaminated milk products (Dietrich and Doherty, 2009).
In the lungs, TB bacilli can reach the pulmonary alveoli where they are phagocytized by resident alveolar macrophages (Kaufmann, 2001). The infected macrophages will invade the adjacent lung epithelium, causing a localized pro-inflammatory response leading to the recruitment of additional monocytes and macrophages from neighboring blood vessels. These recruited cells will in turn be infected with mycobacteria, and will migrate to the lymphoid organs for the establishment of an adaptive immune response, but also can contribute to the dissemination of TB bacilli in the whole body (Russell, 2007).
At the pulmonary epithelium level, the aggregation of macrophages and lymphocytes leads to the formation of a pluricellular structure characteristic of TB known as the granuloma. This structure adopts an organized and stratified architecture: the infected macrophages are located in the center of the granuloma and surrounded by giant multinucleated cells resulting from the fusion of several macrophages. A layer of lymphocytes, predominantly T cells, associated with a fibrous sheath delineates the periphery of the granuloma (Ulrichs and Kaufmann, 2006; Russell et al., 2009).
This step marks the end of the rapid replication phase of bacteria in the host and the evolution to a so-called « containment » phase during which the disease does not clinically manifest and the host is not contagious. At this stage, the granuloma is highly vascularized and the cells of the immune system are actively recruited to the granuloma. In the center of the granuloma, the intervention of the immune system cells will cause necrosis of infected macrophages, leading to the formation of a characteristic necrotic center, the caseum. At a later stage, the granuloma develops a more marked fibrous sheath and the number of blood vessels that penetrate the structure decreases considerably; the center of the granuloma then becomes hypoxic. In this caseous granuloma, bacteria can persist for many years. This stage is known as latent tuberculosis infection (LTBI) (Figure 4) (Ulrichs and Kaufmann, 2006; Russell et al., 2009).
In 90% of the infected individuals, the immune system will successfully keep the bacilli dormant at this stage and prevent the development of the disease. In the remaining 10% of individuals, failure of the “containment” phase will occur, also possibly favored by various factors affecting the patient’s immune system, such co-infection with HIV, malnutrition or diabetes, or tobacco use. As a consequence, the granuloma caseates, breaks, and spills the infectious bacilli into the respiratory system of the patients. The patients will develop active TB and become contagious (Russell, 2007). For patients who do not suffer from immuno-deficiency, half of the total risk of progressing to active TB over the lifetime is concentrated over the first 2-3 years after primo-infection (Blower et al., 1995). Such time window is therefore often used as a study period in molecular epidemiological studies to detect potential recent transmission of TB in a patient population.
Table of contents :
Introduction
A. Tuberculosis: A brief historical perspective
B. The mycobacteria
C. M. tuberculosis genome, evolution and phylogeny
D. Tuberculosis pathology and infection cycle
E. Global tuberculosis epidemiology
F. Tuberculosis treatment and drug resistanc
1. First line treatment
2. Drug resistance and second line treatmen
3. Drug resistance mechanisms and targets
a. First line anti-TB drugs 18 b. Second-line anti-TB drugs
G. Epidemiology of MDR and XDR TB
H. TB situation in Lebanon and impact of the Syrian crisis
I. Diagnosis
1. Immunological tests: TST, IGRA
2. Imaging
3. Specimen collection
4. Microscopy
5. Primary culture
6. Phenotypic drug susceptibility testing
7. Classical molecular diagnostics: Nucleic acid amplification tests (NAAT)
a. Gene Xpert MTB/RIF
b. Line probe assays
c. Seegene Anyplex MTB/NTM, MTB/MDR, MTB/XDR
8. Molecular surveillance by conventional genotyping
a. IS6110 RFLP
b. Spoligotyping
c. MIRU-VNTR typing
9. Combined molecular diagnostics and surveillance by next-generation sequencing (NGS)
a. Whole genome sequencing (WGS)
b. Next generation sequencing assays: Next Gen-RDST and DeepleMycTB
Project objectives and design
Methods
A. Nationwide study setup and design
B. Phenotypic and classical molecular testing
1. Microscopic examination
2. GenXpert MTB/RIF
3. Anyplex MTB/NTM, MTB/MDR, MTB/XDR Real-time detection Kit
4. Primary culture
5. Phenotypic DST
C. Advanced molecular testing
1. WGS
2. Targeted deep sequencing using Deeplex-MycTB
D. Conventional genotyping using MIRU-VNTR typing
E. Statistical analysis
Results and Discussion
Part I: Prevalence of drug resistant tuberculosis in Lebanon: A 12-month nationwide study
Part II: Prevalence of drug resistant tuberculosis using Deeplex-MycTB and phenotypic DST, June 2016 – November 2017
Part III: Prevalence of zoonotic TB in Lebanon
General conclusion
References