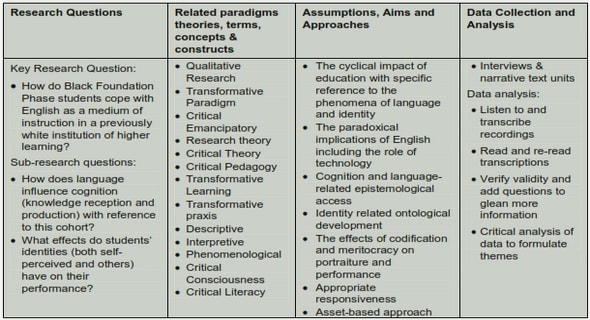
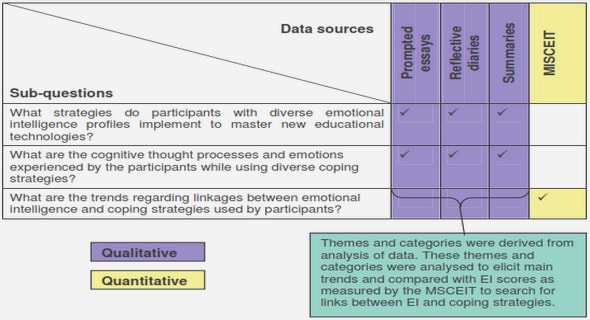
Get Complete Project Material File(s) Now! »
Investigation of amoebapore A pH-dependent activity
Introduction
Amoebapore A dimerisation
The pore-forming activity of APA-1 has been proposed to occur via a pH-dependent dimerisation event, where the protein is inactive in the monomeric form and becomes active when it forms a dimer under slightly acidic conditions [24]. Although no structural description of the dimerisation interface is available, it has been postulated that the ionisation states of four residues are crucial for dimer formation: H75, A63, E2 and K64 [24]. Two salt bridges are formed by these four residues to give an anti-parallel head-to-head dimer. It has been postulated that dimers accumulate on the target membrane and aggregate to form pore structures when a threshold concentration of APA-1 is reached [24]. However, detailed structural information of the dimer and its functionality in APA-1 pore-forming activity remains unsolved. Moreover, it is unclear which ionisable side-chains are functionally important in dimer formation, membrane-binding and pore formation.
NMR spectroscopy to study amoebapore A dimerisation
Identification of residues at the dimer interface
To model the APA-1 dimer structure, residues located at the dimer interface must be identified. Each NMR-active nucleus has a characteristic chemical shift, reflecting its local chemical environment (Section 2.1.3.2). The transition of APA-1 from monomeric to dimeric states should lead to perturbations of the local chemical environments of nuclei located at the dimer interface, thereby leading to chemical shift changes of the corresponding resonances in the NMR spectra [157]. The 2D 1H−15N HSQC spectrum has been frequently used for studying protein-protein interactions [158-162]. By modulating the solution conditions, e.g., pH or protein concentration, to induce APA-1 dimerisation, chemical shift mapping coupled with the backbone assignments can be used to probe the self-association of APA-1, and thus identify the residues located at the dimer interface. The results will be used to guide the design of site-directed mutants that further probe key residues involved in APA-1 activity.
Determination of the pKa values of ionisable groups
Given the pH-dependent activity of APA-1, the ionisation states of the Asp, Glu, His and Lys (no Arg in the APA-1 sequence) residues are likely to play important roles in APA-1 function.
The intrinsic pKa values of these ionisable groups are 3.9, 4.3, 6.5 and 10.4 for Asp, Glu, His and Lys, respectively [43]. Nonetheless, in proteins these pKa values vary over a wide range due to the influence of local environments. Based on the pKa values of ionisable residues assessed in 78 different folded proteins, the values have been found to range between 0.5 and 9.2 for Asp, 2.1 and 8.8 for Glu, 2.4 and 9.2 for His, and 5.7 and 12.1 for Lys residues [48]. The pKa values are influenced by factors such as electrostatic interactions, hydrogen bonding, or the extent of solvent exposure in a protein structure [163-166]. Residues with unusual pKa values often reside at ligand binding or protein interaction sites, or are involved in enzymatic catalytic functions [167-169]. Therefore, the determination of pKa values of ionisable residues can provide insights into the function of these residues in proteins. Figure 3.1 presents the charged residues of APA-1.
Figure 3.1: Ionisable residues of APA-1. (A) Acidic amino acids: the six Asp residues are presented in red and the three Glu residues are in magenta; (B) basic amino acids: the eight Lys are depicted in blue and the sole His is coloured green. Residue assignment information is also provided.
Other biophysical methods
Besides NMR spectroscopy, other methods such as size exclusion chromatography and small angle X-ray scattering can be used to study the APA-1 monomer-dimer equilibrium.
Size exclusion chromatography
Small-zone size exclusion chromatography (SEC) analysis can be used to provide the dimer-monomer equilibrium of APA-1 under different pH values and protein concentrations. However, dilution of the protein concentration (depending on the void volume of the column) and dissociation of the protein complex during passage through the column can influence the population of monomer and dimer species [170] and variable protein retention times [171, 172]. To overcome these problems large-zone SEC can be used. The method requires large protein volumes to saturate an analytical size-exclusion column (usually a few mL volume) and establish a plateau region in which no dilution of the sample occurs. Shifts in the boundary positions of the plateau region as a function of protein concentration can be analysed to examine quantitative information describing the protein self-association equilibrium, such as the fraction of monomer and dimer, and the equilibrium association constant [173, 174]. Unfortunately, because reasonably large sample volumes are required over a range of concentrations (µM to mM), relatively large quantities of protein are needed. Thus, this method was considered to be not suitable for characterising the monomer-dimer equilibrium of APA-1.
Small angle X-ray scattering
Small angle X-ray scattering (SAXS) is a low-resolution technique for characterising gross structural information of protein molecules and protein complexes in solution with sizes ranging from a few kDa to several thousand kDa [175]. The sample solution environment can be easily monitored, therefore making SAXS useful for mapping structural changes to a protein upon environmental perturbations [176, 177]. The basic principle of SAXS is that the incident X-ray beam collides with molecules in solution elastically, leading to the scattering of reflected waves in all directions. The reflected waves interfere with each other, resulting in constructive interference along certain angles and a unique scattering pattern. When small angles are applied, the scattering profiles provide information about the global structure and conformation (e.g., size and shape) of the protein molecule [178]. Figure 3.2 shows a schematic diagram of a basic SAXS experiment.
Figure 3.2: Schematic representation of a SAXS experiment. The diagram is made according to [179]. Incident beam (ki) collides with molecules in solution, scattered with angle 2θ and recorded on the detector. The scattered beam (kr) is equal to 2π/λ. The momentum transfer (Q) is the vector difference between ki and kr. The background scattering profile is subtracted from the sample.
When X-rays with a known wavelength (λ) are scattered from a sample, the scattering vector or momentum transfer (Q), can be calculated using Eq. 3.1 [180]:
where 2θ is the angle between the incident and scattered beam. The scattered intensity, I(Q), is recorded as a function of Q. Since the λ is fixed, I(Q) is essentially dependent on the scattering angle θ. Scattering data from the solvent is subtracted from the scattering profile. SAXS experiments to obtain high-resolution structural information require highly pure, soluble and monodisperse protein samples with sufficient dilution so that no significant attractive or repulsive interactions exist between molecules. All protein molecules in a sample contribute to the SAXS signal, thereby the random positions and orientations of the molecules give rise to an isotropic intensity distribution which reflects the scattering from a single protein molecule averaged over all orientations [181, 182].
The scattering patterns generated from protein molecules in a sample are presented as averaged 1D curves. Structural parameters such as the MW, radius of gyration (Rg) and maximum intramolecular distance (Dmax) of the protein can be determined from the 1D curves, providing a low-resolution overall shape of the protein [183]. The Rg, which provides information of mass distribution within a protein, can be estimated using the Guinier approximation [183]:
where I(0) is the scattered intensity at zero angle which is proportional to the MW of the protein. By plotting In(I(Q)) versus Q2 and fitting the slope and intercept, Rg can be obtained [183]. Small amounts of aggregation of the sample in solution can lead to an upturn in the Guinier plot and give rise to biased Rg and I(0) values. The software GNOM [184] can be used to indirectly Fourier transform the scattering profile to obtain the interatomic distance distribution function, P(r), of the scattering molecules [185]. The P(r) profile can provide the shape and volume occupied by a protein or protein complex and is sensitive to the symmetry and domain structure within a molecule.
In addition, information obtained from a SAXS profile can be used to assess different protein models with defined geometry to estimate the rough shape and size of the protein. Protein models with simple shapes (e.g., sphere, cylinders and ellipsoids) are usually employed as a first step in scattering pattern interpretation. The calculated and the experimental scattering patterns are compared and a close-to-native solution structure from different models of the protein can be achieved [186]. Moreover, the approximate 3D structure of a protein such as size and shape can be reconstructed from the 1D scattering data using ab initio analysis, assuming a homogenous solution and constant scattering density within the molecules. For example, programmes DAMMIN [187] and DAMMIF [188] are able to reconstruct the overall shape of a protein molecule by finite volume elements (i.e., dummy atoms) to fit the experimental data. Both programmes use simulated annealing to construct compact and interconnected models. GASBOR [189] can be used to reconstruct proteins structures by a chain-like ensemble of dummy residues. These dummy atom/residue model approaches allow several models to be generated which may fit equally well to the experimental SAXS data. To reduce ambiguities, DAMAVER [190] is used to place the models into structurally similar classes and generate an average structure from each class. The generated ab initio model can be superimposed with the atomic structure of the target protein using SUPCOMB [191]. In addition, programmes CRYSOL [192] and AXES [193] are modelling tools that directly operate on known protein atomic models (entered into the protein database) to generate theoretical scattering profiles. The theoretical profiles are evaluated against the experimental profiles to validate the atom model, i.e., least-squares type fitting of the experimental data.
Furthermore, SASREF [194] can be used to perform quaternary structure modelling of a molecular complex with a known protein structure against the experimental scattering data; multiple non-unique structures may be obtained because one or more molecules are free to move in space.
Design of amoebapore A mutants
Site-directed mutagenesis is a useful tool to verify the proposed APA-1 dimer model and examine the importance of the residues that are proposed to be located at the dimer interface. Based on the previously proposed dimer model [24] (Figure 3.3), five mutants were designed by our collaborator in Germany (Prof. Joachim Grötzinger, Department of Biochemistry, Christian-Albrechts-Universität zu Kiel, Medical Faculty, Kiel, Germany). These mutants are proposed to disrupt or remove the pH-dependency of dimer formation, including E2Q, D63N, D64K, H75Q and H75K, where E2Q, D63N, K64Q and H75Q are considered to abolish the ionic interactions of the dimer, and H75K is proposed to eliminate the pH-dependency of dimer formation (at least over pH values 8). Based on NMR perturbation analysis which will be described in Section 3.3.2, additional mutants which are postulated to disrupt or stabilise APA-1 activity were designed (i.e., E2A, K37A, K37Q, F41A and E2Q-K37Q). Biological assay tests along with NMR spectroscopy can be used to probe the activity and fold of these mutants.
Figure 3.3: Proposed APA-1 dimer model in solution. Two ion pairs are involved in the dimer interface, H75D63 and K64E2. Five mutants: E2Q, D63N, D64K, H75Q and H75K, were designed based on this model, which are proposed to either disrupt or stabilise dimer formation.
Aims
The main research aims in this chapter were to:
(1) Use NMR chemical shift perturbation analysis to identify residues located at the dimer interface;
(2) Synthesise selectively 13C/15N-labelled APA-1 (Glu, Asp, Lys and His are labelled) samples to measure side-chain pKa values using NMR spectroscopy;
(3) Perform concentration-dependent chemical shift mapping combined with small-zone SEC to estimate the dissociation constant of the APA-1 monomer-dimer equilibrium;
(4) Perform SAXS experiments to investigate the gross shape and size of APA-1 in solution at different protein concentrations and pH values;
(5) Design and synthesise APA-1 mutants based on the proposed dimer models in order to determine which model is correct, and investigate the importance of dimerisation in APA-1 functionality and identify residues that are crucial for APA-1 activity.
Methods and Materials
Sample preparation
Uniformly 15N-labelled and selectively 13C/15N-labelled APA-1 samples were produced from the cell-free protein expression system and purified as described in Chapter 2. To reduce the cost of using double labelled material and minimise peak overlap, only charged amino acids were 13C/15N-labelled. Potassium glutarate replaced potassium glutamate to ensure sufficient Glu labelling [146].
NMR spectroscopy
APA-1 samples were prepared as described in Chapter 2. NMR experiments were performed at 25 °C. The pH values of the samples were measured before and after each experiment. The average readings were taken for data analysis. The pH showed little variation at most pH values (±0.05 pH unit). However, the pH was not stable above 8.5, where the solution was not buffered. To ensure stable pH values, a 30 min incubation period was emplyed after adjusting the pH, and the pH values were measured again before each NMR experiment.
pH- and concentration-dependent chemical shift mapping
The 1D 1H and 2D 1H−15N HSQC spectra of uniformly 13C/15N-labelled APA-1 samples with concentrations of 210 μM were recorded as a function of pH between 3.0 and 8.5 at 0.5 pH unit increments. The MWs of APA-1 at each pH value were estimated by performing a 1D experiment, as described in Section 2.2.12.1 (Eq. 2.2 and 2.3). The errors of the T2 values were estimated by taking the signal-to-noise ratio:
where A and B are the delay periods used in the 1D spectra, S/N0.25ms and S/N2.9ms are the signal to noise peak intensity ratios recorded using spin-echo delay periods of 0.25 and 2.9 ms in the two 1D spectra, respectively.
The 2D 1H−15N HSQC backbone assignments obtained from Chapter 2 at pH 3.0 were used as an initial starting point for the titration. To investigate the pH and concentration effects on dimer formation, 2D 1H−15N HSQC spectra of uniformly 13C/15N-labelled APA-1 samples were acquired at pH 3.0 and 5.2, and at protein concentrations of 25 and 560 μM (i.e., four samples). Chemical shift changes of the spectra recorded at pH 3.0 and 5.2 at each protein concentration were compared using Eq. 3.4:
where δH and δN represent the 1H and 15N chemical shift differences of a resonance, recorded at two pH values and one of the two concentrations. One standard deviation above the mean of the weighted chemical shift perturbation ( δNH ) for all the resonances was used as a threshold, in which, any residue with a δNH value greater than the threshold value was considered to be influenced over the pH range studied. To separate the pH and concentration effects on the chemical shifts, the δNH obtained at the high and low protein concentrations were then compared to each other. The difference in the absolute weighted chemical shift perturbation values (| δ|) was extracted by subtracting the pH-dependent chemical shift differences for the two protein concentrations [157]
Chapter 1: Introduction of amoebapore A, an antimicrobial protein produced by Entamoeba histolytica
1.1 Introduction of Entamoeba histolytica
1.2 Introduction of amoebapore A
1.3 Structural and biophysical studies of amoebapore A
1.4 Research Aims
Chapter 2: Cell-free production and structural and functional characterisation of recombinant amoebapore A
2.1 Introduction
2.2 Methods and Materials
2.3 Results
2.4.1 Optimisation and troubleshooting during cell-free synthesis of amoebapore A
Chapter 3: Investigation of amoebapore A pH-dependent activity
3.1 Introduction
3.2 Methods and Materials
3.3 Results
3.4 Discussion
Chapter 4: Interaction of amoebapore A with model membranes
4.1 Introduction
4.2 Methods and Materials
4.3 Results
4.4 Discussion
Chapter 5: Conclusion
5.1 Mechanism of amoebapore A dimerisation
5.2 Amoebapore A interaction with model membranes
Appendices
GET THE COMPLETE PROJECT